The Hidden World of Microbiomes and Their Impact on Our Lives
Microbiomes are the diverse communities of microorganisms that inhabit different parts of our bodies, as well as the environment around us. In recent years, research has revealed the vast and complex hidden world of microbiomes and their impact on our lives, from influencing our digestion and immune system to potentially affecting our mood and behavior. Advances in technology have enabled scientists to study microbiomes in unprecedented detail, leading to new insights into their diversity and functions. Understanding the microbiome and its role in human health and disease has the potential to transform how we approach medicine, nutrition, and the environment.

With high diversity, you also get a combination of characters, the human microbiome is consequently no stranger to the good, the bad and the ugly.
There are good microorganisms, then nasty ones, and then good ones that might turn into bad ones.
One of the most famous good/bad bacteria is Staphylococcus aureus commonly known as Staph. It’s generally found on the skin and in the nasal passages of healthy individuals, where it can play a beneficial role in preventing colonization by other, potentially harmful bacteria. However, S. aureus can also cause a range of infections, including skin infections, pneumonia, bloodstream infections, and heart infections. Some strains of S. aureus are antibiotic-resistant, making them particularly difficult to treat. Thus, understanding what triggers the switch from a peaceful commensal bacterium inhabiting our noses to a virulent pathogen is key to identifying potential therapeutic targets.
A study by Wittekind et al. (2022) provided further insight into the mechanisms behind the expression of virulence genes in S. aureus. The research describes the discovery of a novel protein, ScrA (which stands for S. aureus clumping regulator A), in Staphylococcus aureus (SaeRS). ScrA interacts with the SaeRS two-component system (TCS), which is known to regulate the expression of virulence genes in S. aureus. The results show that ScrA plays a key role in the regulation of virulence gene expression by the SaeRS system, and that deletion of the ScrA gene results in a significant decrease in virulence in a mouse infection model. Thus, ScrA could be a promising target for the development of new therapies to treat S. aureus infections.
One of the key methods in Wittekind et al. (2022) experiment was RNA-sequencing to get a glimpse of the gene expression profile of S. aureus. The global view provided by RNA-Seq helped pinpoint one of the S. aureus two-component systems that showed higher expression when ScrA was overexpressed.
Since rRNA accounts for 80-90% of the transcriptome limiting the detection efficiency of desired RNAs by RNA-Seq. The removal of ribosomal RNA (rRNA) before RNA-Seq greatly improves and economizes RNA-Seq. In this study, ribosomal RNA depletion was performed using the Staphylococcus aureus– specific riboPOOL rRNA removal kit.

A Brief Interview with Dr. Marcus Wittekind
To have further insight into the process, challenges of studying human microbiomes, and the most interesting findings related to small RNAs (sRNAs) we interviewed Dr. Marcus Wittekind.
Marcus is a research scientist at Ohio University and is a member of Dr. Ronan Carroll’s Lab. His research is focused on bacterial pathogenesis and the role RNA molecules play in the bacterial cell. Meet one of the scientists behind the research:
- What inspired you to pursue research on human microbiomes?
I have always had an interest in how microbes interact with their host. Staphylococcus aureus is particularly interesting to me in that it is found in ~30% of the population as a human commensal and just sits in the nose without any issues. Yet, when S. aureus migrates to other areas you can get devastating disease. It’s fascinating how S. aureus is able to make this transition and switch from a relatively passive existence to a virulent pathogen. Along with S. aureus, it’s astounding how little we actually know about the microbiome and how it influences our health. It’s exciting to live during a time when we’re uncovering these connections.
- What are the most interesting findings from your latest research on the commensal bacteria Staphylococcus aureus?
My findings about S. aureus have focused primarily on a single small protein ScrA. Although my research has been focused on a single protein, I think it can serve as an example of just how much we have left to learn. I found ScrA to act as a sort of link between two well-studied regulatory systems in S. aureus. While this is an interesting subject in its own right, I think where this story comes from is particularly interesting. My mentor Ronan Carroll originally identified the scrA gene, which was at the time called tsr37, as a small non-coding RNA. However, we later came to find out that some of these small RNAs actually encoded small proteins. Now this isn’t surprising, we already know of a toxin encoded on a small RNA. However, it makes me wonder how many more proteins are we overlooking as being just small RNAs? Some of my studies also suggest that ScrA is really only important when S. aureus is infecting the heart. In laboratory conditions we don’t really see any changes when we delete scrA, which would normally lead to us just moving on without discerning the function of ScrA. Only due to marked phenotypes when we overexpress ScrA did we even become interested in its function. How many more genes play a vital role in virulence but are being overlooked because we can’t see anything in the lab? I think ScrA serves as a reminder of how unassuming genes can actually have a larger role than what we see on the benchtop.
- What are some of the biggest challenges researchers face in the field of microbiomes?
The sheer complexity of the interactions between pathogens and their host. For me, this has manifested as finding the exact conditions in which ScrA is activated and carries out its function. All I really know is that scrA plays a role in infecting the heart. However, the question still remains as to what triggers scrA production. Nutrient abundance? Immune system components? Temperature? Host signals? At this point, I can only guess. For me I only have to focus on a single organism. The complexity drastically increases when you consider environments with multiple organisms such as the digestive system, skin, or wounds. While the complexity is fascinating it is also difficult to wrap your head around exactly what is taking place.
- What technologies and methods are key for your research?
There are many different technologies and methods that are essential for my work. However, a few stand out to me. I went into this project with no idea what was causing the phenotypes. So, we decided to cast a wide net and see what was being altered in the cell. RNA-sequencing actually gave us our first hint of what was going on. We saw global changes in gene expression; however, we were able to pick out one system in particular that showed promise. One of the two-component systems in S. aureus (SaeRS) showed higher expression when we overexpressed ScrA. Thanks to the global view we can get by using RNA-seq we were able to identify a potential mechanism with one experiment as opposed to screening individual regulators.
On the same note, mass spectrometry allowed us to get a global view of protein changes. This was particularly useful when we were identifying what host factors were being bound when we overexpress or delete scrA. We were able to “shave” the surface of the cells with immobilized trypsin and identify the exact proteins present, and more importantly what proteins could be accessed by the trypsin. Being able to quickly sort through all the different components was essential to forming a working model for ScrA mediated aggregation.
Finally, we can’t ignore how essential animal models are for studying virulence. While it would be great and I look forward to a day when we no longer need to perform animal experiments, right now they are absolutely vital to understanding these pathogens. We utilized a mouse model of systemic infection to determine if scrA was essential for virulence. Not only was I able to show that scrA is needed for virulence, but I was also able to show that scrA is primarily needed for heart infections. This is something we wouldn’t have known without animal models. When we delete scrA and use it in our in vitro experiments, we see limited effects and only under specific conditions. However, we saw a drastic decrease in virulence in a mouse model.
- What are some potential applications of your research on human health?
One of the primary reasons I want to understand S. aureus virulence is to identify potential therapeutic targets. It’s well known that antibiotic resistance is on the rise and at some point, we are going to need alternative treatments. S. aureus is interesting because in most cases it just sits in the nose and doesn’t cause disease. If we can understand what triggers that switch from a passive carry to an aggressive infection, we might be able to force S. aureus to stay in a passive state or at least limit its virulence. I’ve shown ScrA is needed for effective heart infection by S. aureus. It may be possible to target ScrA and inactivate it, reducing its ability to infect the heart. This could be useful in people undergoing heart surgeries, especially in cases with indwelling medical devices, which may introduce S. aureus into the heart.
- What advice would you give to someone interested in pursuing a career in Bacteriology?
Bacteriology is a wide field, take your time to explore different aspects and find something that really interests you. The sheer volume of information can be overwhelming when you get started, but as time goes on it becomes more familiar. The best way to see what really interests you is to get involved in research. Reach out to people whose research interests you and find opportunities to get involved. I know how intimidating this idea can be (I started researching as an undergraduate) but many professors are happy to have interested people join their lab regardless of experience. Most importantly don’t feel obligated to stick with the first thing you start studying. One of the things I love about bacteriology is how much there is to learn. If you don’t like what you’re studying, there is always something else you can try. It’s important to find your niche and what you enjoy. Being passionate about your work is an important part of this field.
Biocabulary:
Two-component systems (TCSs) are signaling pathways that allow bacteria to sense and respond to changes in their environment. A TCS consists of two proteins: a sensor histidine kinase and a response regulator. The sensor histidine kinase detects a specific environmental signal and transfers a phosphate group to the response regulator protein, which then activates or represses the expression of specific genes.
Small RNAs (sRNAs) are short, non-coding RNA molecules that typically range in size from 50 to 500 nucleotides. They are important regulators of gene expression in bacteria, archaea, and eukaryotes, and play diverse roles in cellular processes such as stress response, metabolism, and virulence.


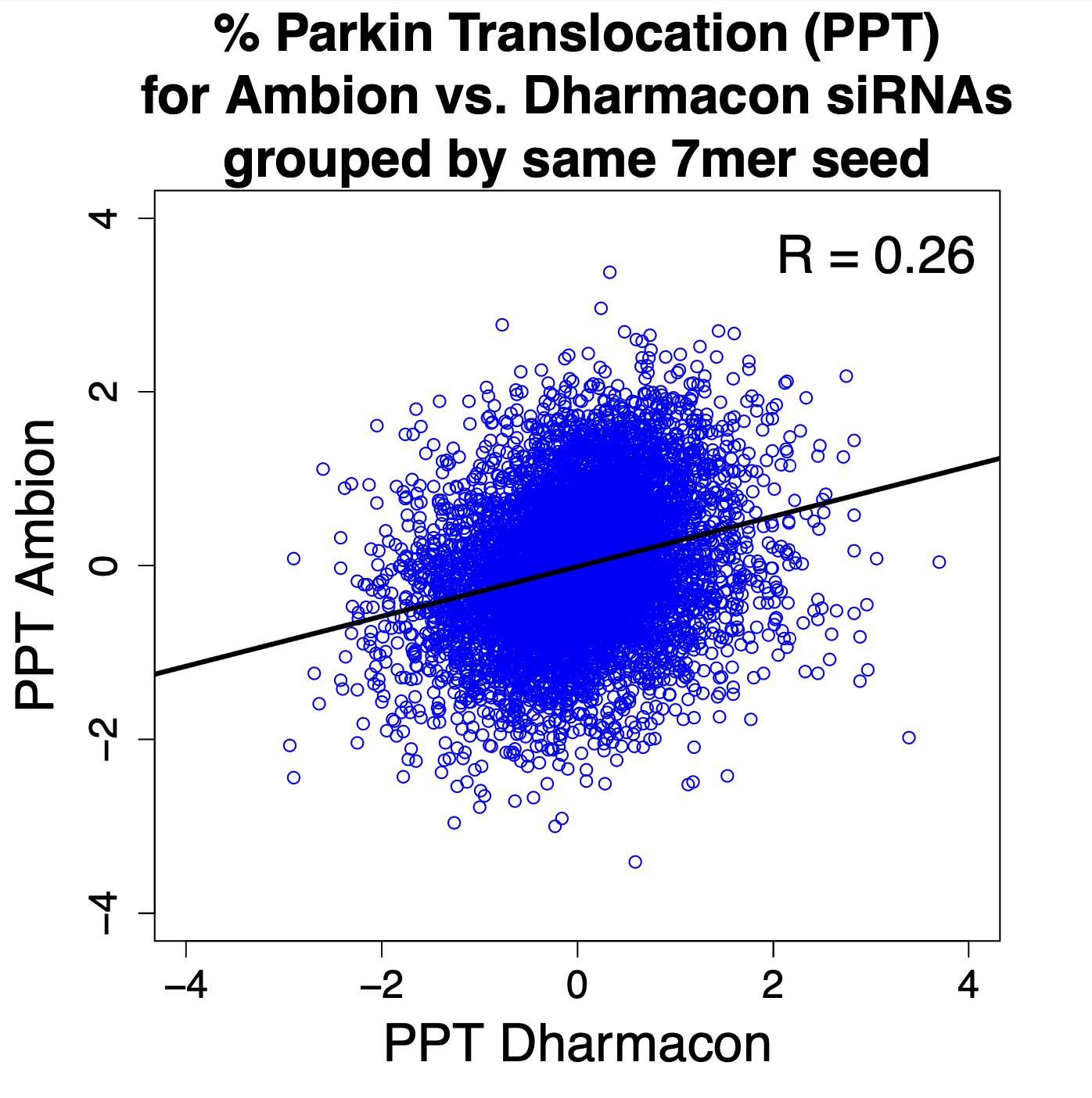
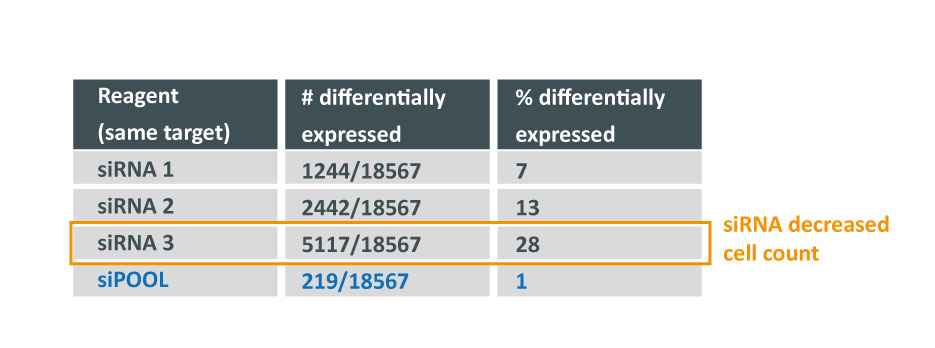
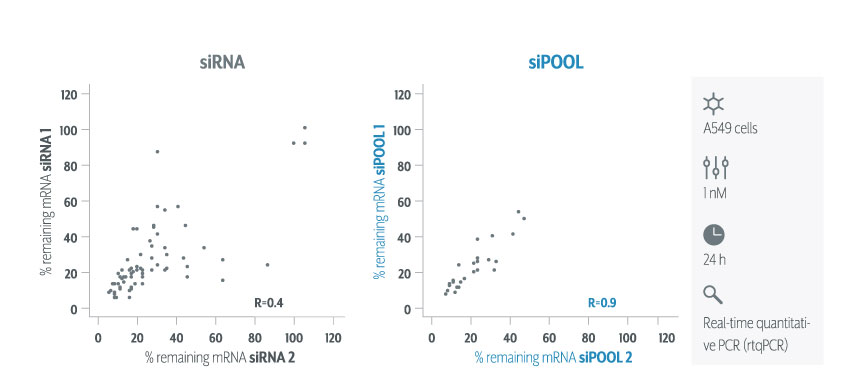
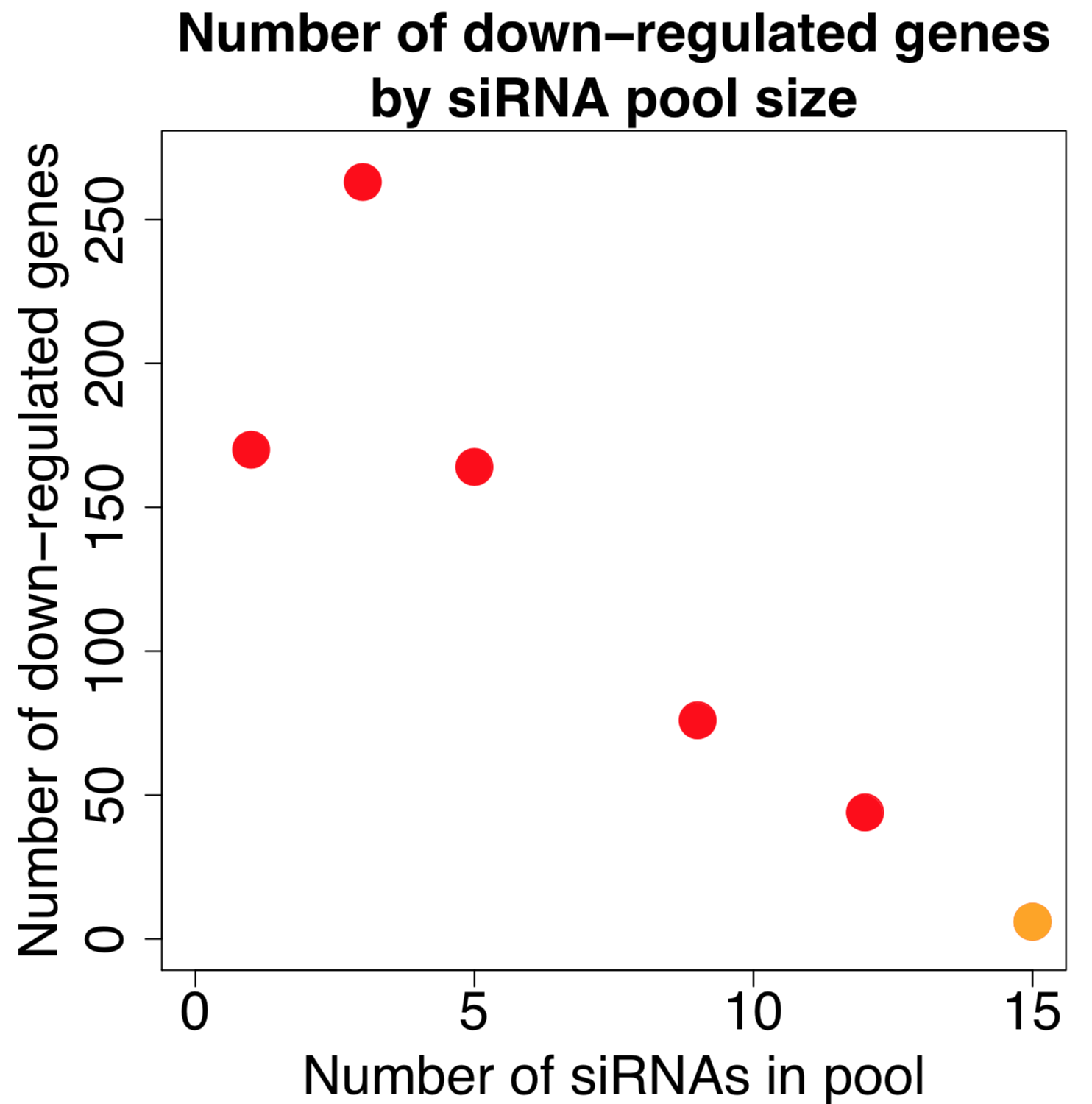
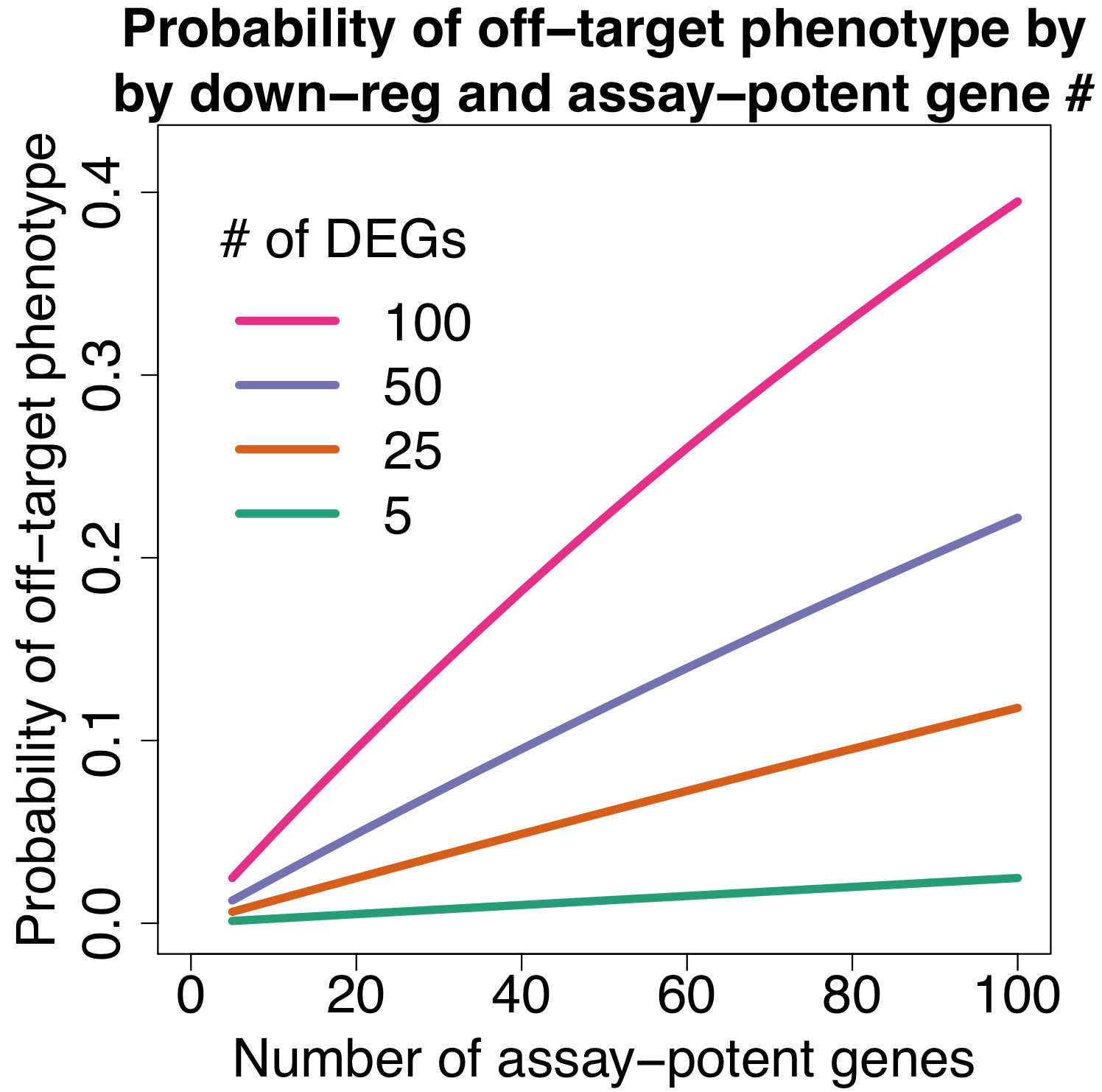
 for Ambion vs. Dharmacon siRNAs grouped by same 7mer seed")