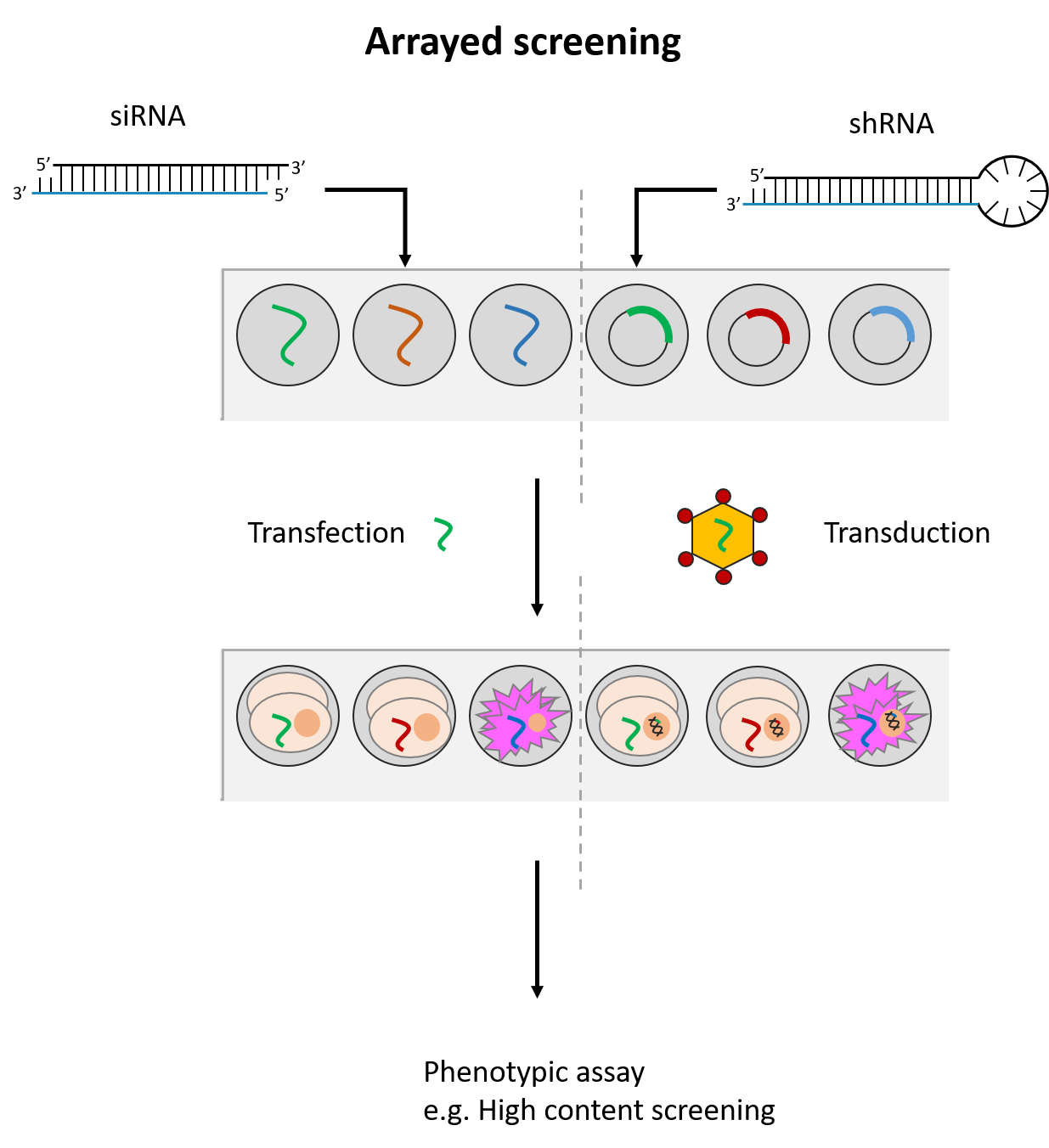
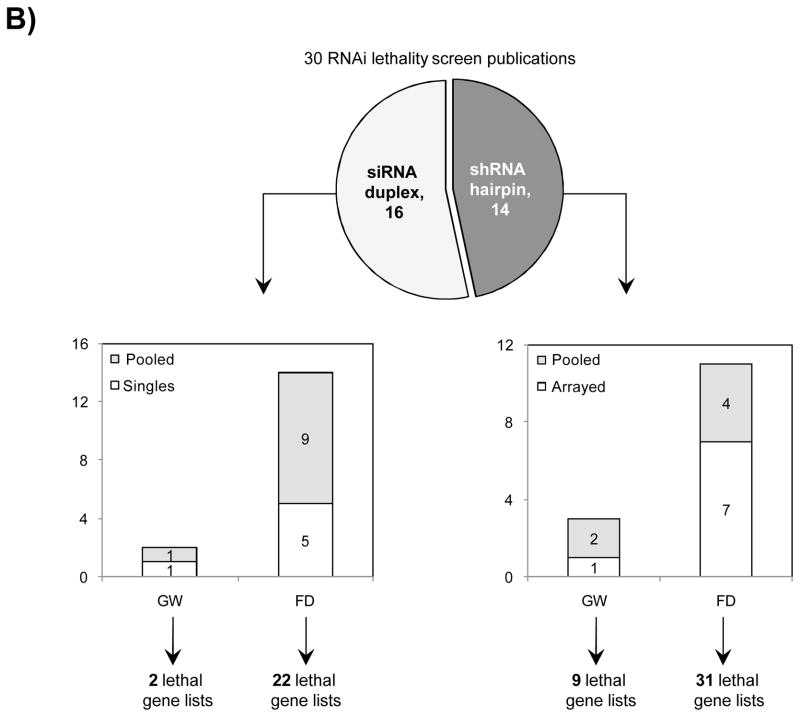
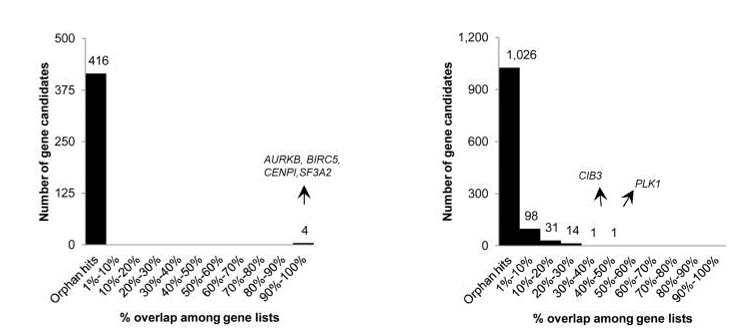
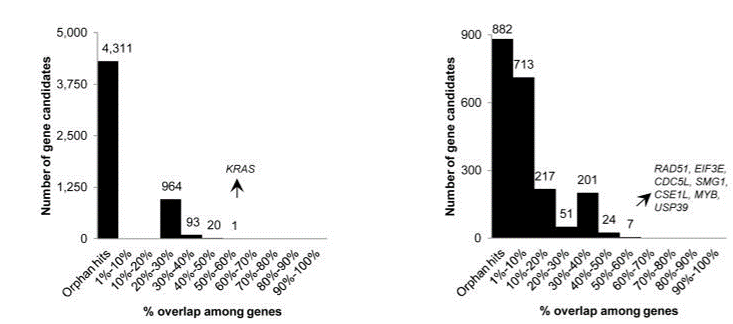
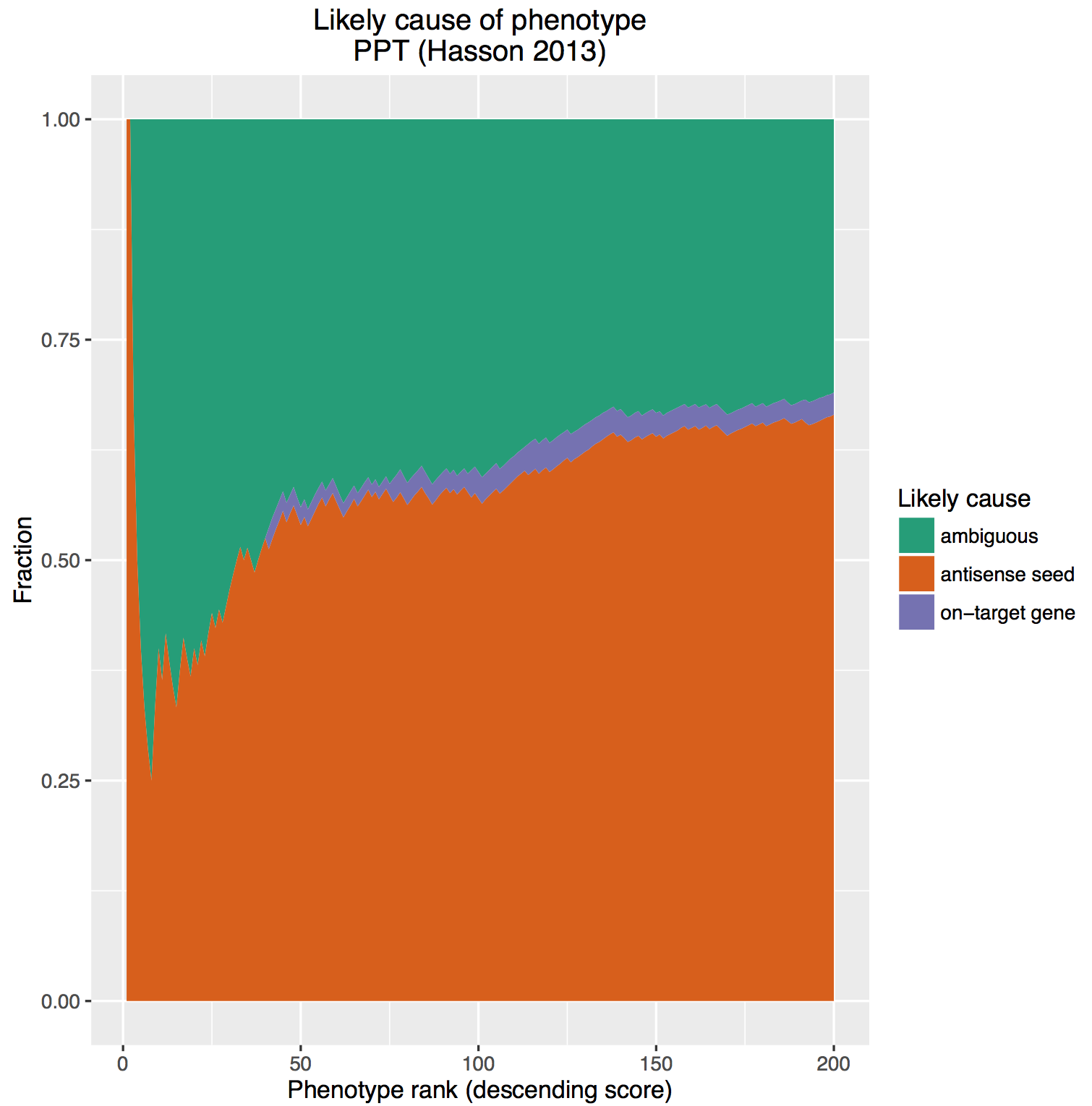
CRISPR is a gene editing technique based on tools and principles learnt from the bacterial immune system. Gaining immense popularity world-wide, many are trying to establish CRISPR in their favourite model systems to study gene function. Here, we highlight issues to be aware of when using CRISPR and what one can do to counter or manage them.
To simplify matters, we have classified what could go wrong while performing CRISPR into three main categories, accompanied by associated exclamations one may hear in the process:
- “Hmm… I don’t see anything.” – Absence of phenotype
- “This is taking wayyy too long.” – Inefficient editing
- “What the *@#?!” – Unexpected phenotypes
First, some key terms…
Cas9: The bacterial RNA-guided endonuclease that mediates cutting of the DNA. The most commonly used Cas9 ortholog is from Streptococcus Pyogenes and can be introduced into cells in the form of DNA, mRNA, or protein.
sgRNA: single guide RNA composed of a 17-20 base long guide RNA (gRNA) which hybridizes to its complementary DNA sequence on the genome, defining the target site. This is often joined to a ~70-80 base long transactivating crRNA (tracrRNA), a constant region that mediates recruitment of Cas9. sgRNAs can be introduced as one unit or in its separate components – gRNA and tracRNA – as DNA or RNA.
PAM: protospacer adjacent motif, a trinucleotide sequence 3’ adjacent to the gene editing site required for Cas9 to bind and mediate cleavage. Sequence is NGG for Cas9 from Streptococcus Pyogenes though NAG is often recognized as well. PAM sequences differ between various forms of Cas enzymes.
- “Hmm… I don’t see anything.” – Absence of phenotype
The anti-climax of a null result may stem from adaptation where the cell or organism alters other gene pathways to compensate for the loss-of-function of the target gene.
This problem is most visible to those maintaining Drosophila stocks as strength of phenotype typically decreases over multiple generations. The phenomenon is also well-documented in other models such as yeast (Teng X et al., 2013), zebrafish (Rossi et al., 2016, covered in a previous blogpost) and mice (Babaric et al., 2007). A notable Developmental Cell paper recently reported adaptation in cells (Cerikan et al., 2016) where prolonged knock-down (KD) or knock-out (KO) yielded no visible phenotype as opposed to acute KD by RNAi.
Multiple cell passages increase genetic drift, providing opportunities for the system to adapt to counter the disruptive effects of a gene knock-out. It is therefore prudent to preserve early passages of clones during clonal selection and limit multiple passages prior to assay measurement.
Besides adaptation, redundancy may also account for an absence of phenotype. Paralogous genes (i.e. genes closely related in structure or function) often exist in model systems that can fully or partially compensate for the loss-of-function of the target gene. About 50% of mouse genes and at least 17% of human genes have paralogues that may mask loss-of-function phenotypes.
One can find paralogous genes arising from gene duplication with this database and by checking existing literature. If they do exist, a co-knock-out/knock-down approach may be necessary.
- “This is taking wayyy too long.” – Inefficient editing
Despite the high efficiency of Cas9-mediated cleavage, obtaining the desired gene knock-out can still be a tedious and time-consuming process, with wide-ranging overall efficiencies of 1-79% (Unniyampurath et al., 2016).
These challenges often stem from issues associated with the cell line of choice. Due to many standard cell lines being polyploid (containing multiple copies of chromosomes), every copy of the gene has to be disrupted to ensure a complete knock-out. A process aggravated by the need for a homozygous knock-out. Transfection efficiencies, how well the cell line tolerates clonal selection and the impact of the gene modification on cell viability can also impact outcomes. If performing homology directed repair (HDR) to introduce a new sequence at the cut site, clone screening efforts have to be amplified due to the lower frequency of HDR events compared to indels.
Understanding the characteristics of your cell line and ensuring sufficient numbers of clones are screened is essential to avoid mindless weeks repeating experiments!
Editing efficiency may also be hindered by genomic accessibility. gRNAs targeting transcriptional start sites or promoters were found to be more efficient than intergenic sites due to the open chromatin structure in these areas (Liu X et al., 2016). Numerous design criteria have been recommended to ensure high cutting efficiency but performance of gRNAs may still vary. Therefore it is advisable to use at least 3 sgRNAs per gene to increase chances of success.
Sidenote: Looking for someone who can design CRISPR sgRNAs for you? siTOOLs Biotech’s CRISPR sgRNA design service couples our long-standing experience in off-target filtering with published gRNA design criterion to generate reliable gRNA sequences. Send us your enquiry and we will get back to you.
- “What the *@#?!” – Unexpected phenotypes
Unexpected results can stem from off-target effects or in some cases, may be a real effect that requires some brain rattling to make sense of.
Off-target effects are still a cause of concern for CRISPR and vary widely with different gRNA sequences ranging from 0 to up to 150 in one report (Tsai et al., 2015). In another study, ~10 to > 1000 off-target binding sites were found that varied with sgRNA sequence (Kuscu et al., 2014).
Toxicity correlated with increased off-targeting (Morgens et al., 2017) and the use of safe-targeting controls (i.e. where gRNAs are directed towards sites where cleavage is predicted to have minimal impact) was recommended. This served as a more appropriate measure of nuclease-induced toxicity as opposed to non-targeting controls that might not lead to cleavage.
Some other strategies to minimize off-targets:
- Use the Cas9 recombinant protein/mRNA rather than a plasmid or keep DNA transfection amounts low (plasmid-driven prolonged Cas9 expression increased off-targeting events as reported by Liang et al., 2015)
- Use truncated gRNAs of 17-18 nucleotides
- Use D10A Cas9 nickase and paired gRNAs
- Use a Cas9 ortholog with a longer PAM requirement
Despite our efforts to predict off-target effects, two reported sources of potential off-targets make prediction challenging:
a) Single nucleotide variants from clonal heterogeneity
b) Cas9 effects on mRNA translation
a) Single nucleotide variants from clonal heterogeneity
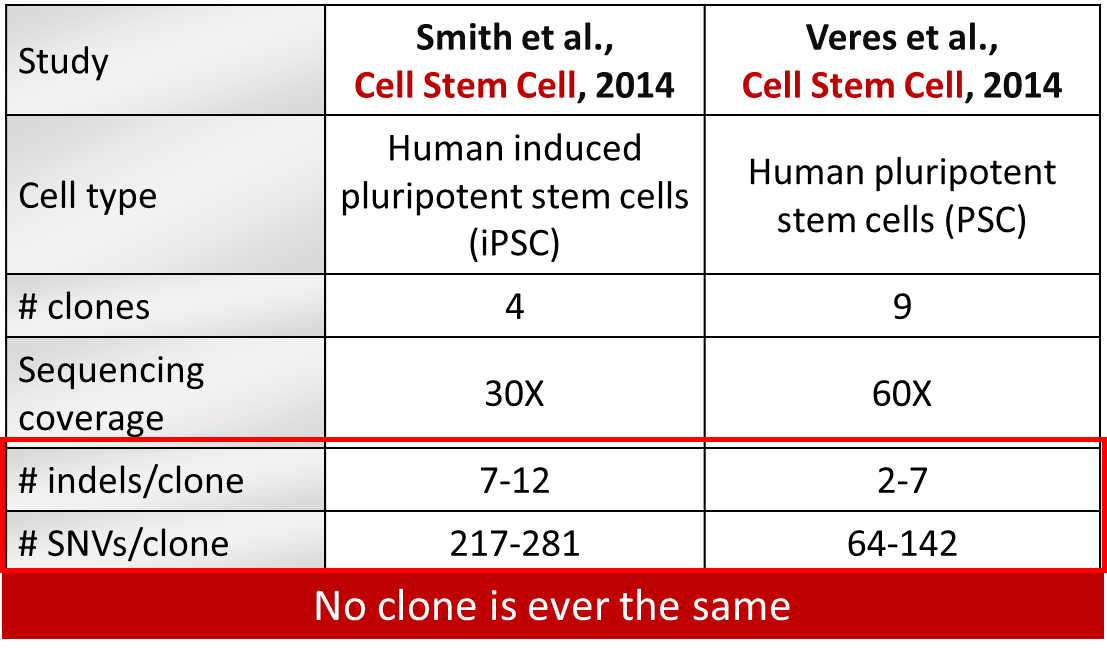
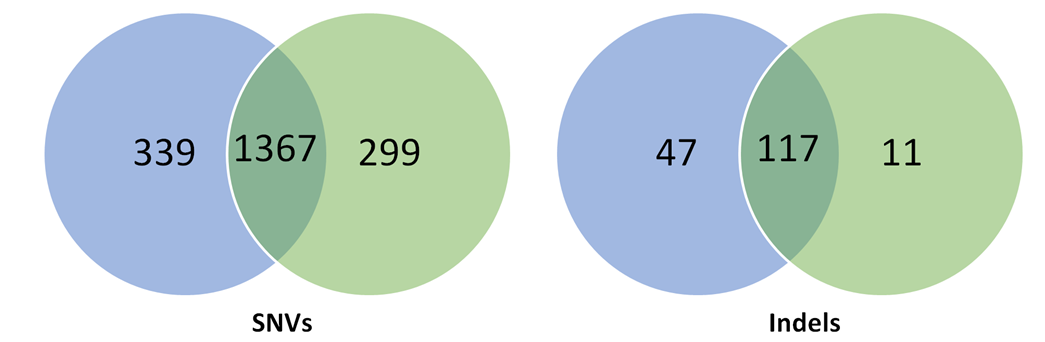
Table 1: Spontaneous SNVs and indels generated over clonal selection in human pluripotent stem cells.
Two studies (Smith et al., 2014; Veres et al., 2014) carried out in pluripotent stem cells to detect off-targets saw a higher specificity of Cas9 in these cells compared to cancer cell lines but shockingly, rather large clonal heterogeneity (Table 1). Each clone generated from the parental cell line had on average 100 unique SNVs per clone and 2-5 indels not induced by the gene modification but arising spontaneously during cell culture.
| Target and off-target indel frequencies |
| Number of mismatches |
Number of genomic sites |
Cas9 targeting efficiency |
| 0 |
1 |
53.9% |
| 1 |
0 |
– |
| 2 |
0 → 1 |
36.7% |
| 3 |
32 |
~0.15% per site |
Table 2: Editing efficiencies at off-target sites with 0-3 mismatches. Condition of SNV enhancing editing efficiency shown in bold.
Yang et al., 2014 then goes on to demonstrate how an SNV at the wrong place at the wrong time can produce a high-efficiency off-target site. The said SNV corrected a mismatch at an off-target site, reducing mismatch number from 3 to 2, which increased Cas9 –mediated indel frequency to ~37%!
To manage clonal heterogeneity, we recommend performing deep sequencing to fully characterize the knock-out clone and its parental wild-type cell line. Once the locations of SNVs are identified, these can be aligned with potential off-target gRNA binding sites to check for interference. Check locations of identified unique SNVs or indels to see if they are impacting genes that may play a relevant role in your studied phenotype.
b) Cas9 effects on mRNA translation
A Scientific Reports study (Liu Y et al., 2016) reported a worrying finding that Cas9 could be recruited by gRNAs to mRNAs and block their translation. Neither PAM sequences nor Cas9 enzyme activity was required for this and the effect varied with gRNA sequence. Cas9-mediated mRNA translation suppression produced a 30-60% decrease in protein levels, sufficient to impact downstream phenotypes. For example, a gRNA targeting VEGFA with an off-target binding site to the mRNA of oncogene, B3GNT8, produced a nearly 50% drop in B3GNT8 protein levels with a corresponding drop in cell viability. This was partially rescued by overexpressing B3GNT8 with a vector.
It is still unclear to what extent this phenomenon occurs. There have been limited reports on this mechanism so far, but if true, would have a far-ranging impact. The study found gRNAs with single base mismatches at position 8-20 were still able to carry out Cas9-mediated translation repression. This low hybridization stringency requirement would make off-targets impossible to predict.
CRISPR is no doubt a powerful technology, but it still brings many unknowns. After its discovery in the 1990s, RNAi experienced a similar exponential uptake and use by the scientific community. It took several years for the problem of siRNA off-targets to become visible. Unfortunately by that time, enormous resources and energy had been sunk into large RNAi screens, which yielded numerous false hits and difficult-to-interpret data.
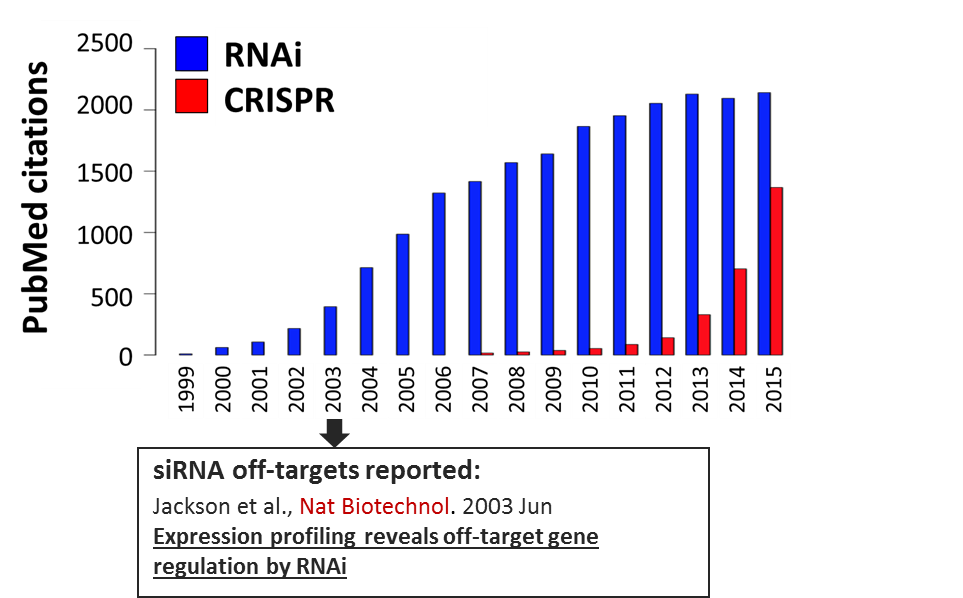
Figure 1. Pubmed Citations (1999-2015) with CRISPR or RNAi in Title/Abstract/Summary
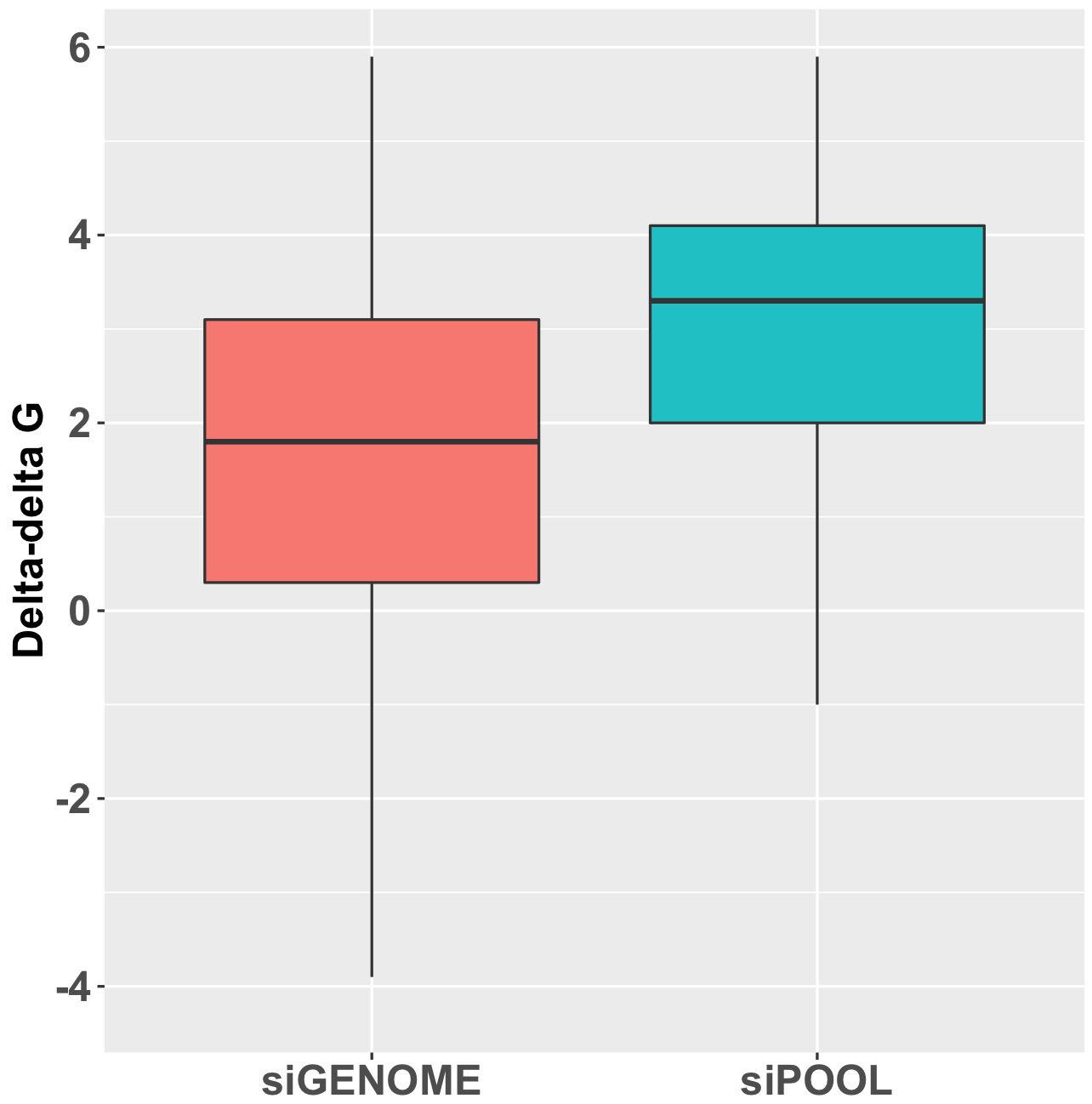
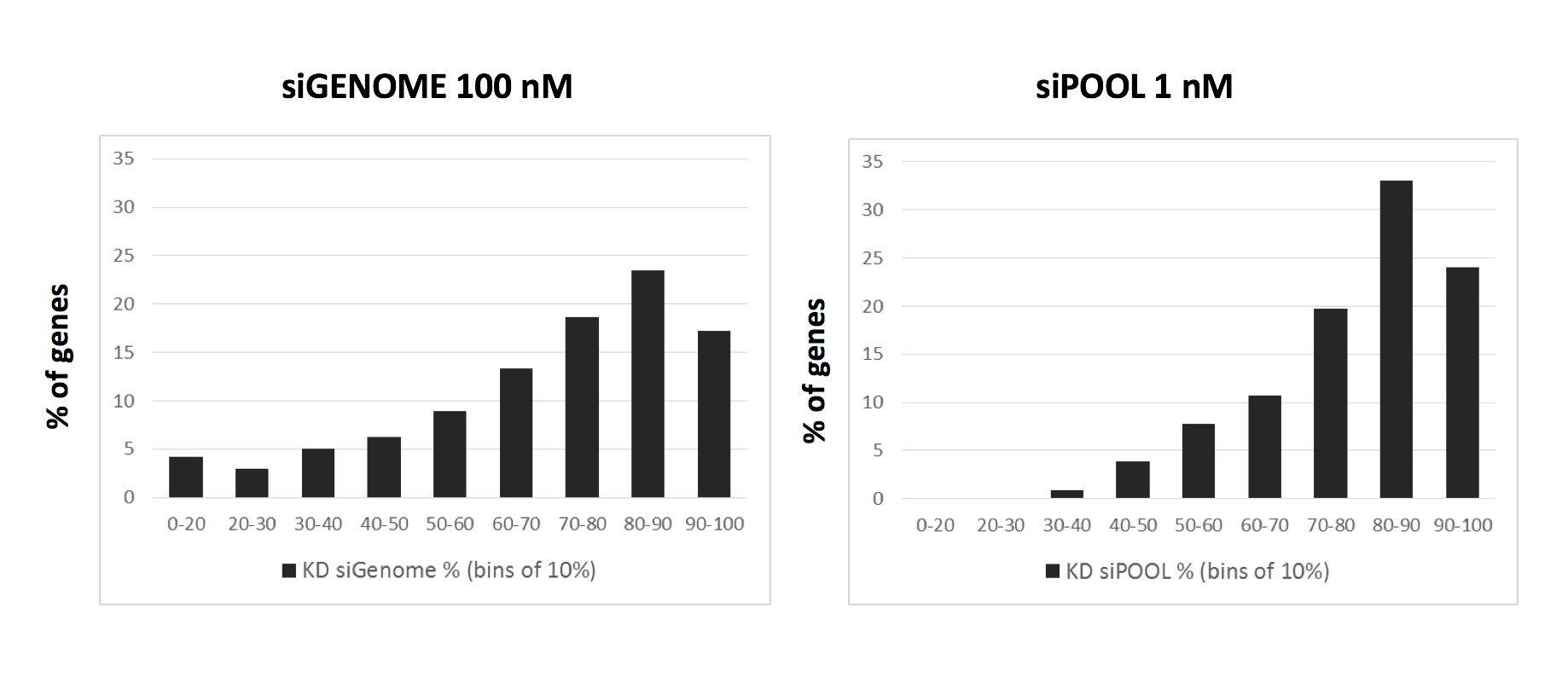
Thankfully we now have siPOOLs, or high-complexity defined siRNA pools (from siTOOLs Biotech). These custom-designed pools of 30 unique siRNAs counter the off-target effects often seen with single siRNAs or low complexity siRNA pools of 3-4 siRNAs (Marine et al., 2012, Hannus et al., 2014). Efficient at 1 nM in standard cell lines, it is the optimal RNAi reagent for highly specific, efficient and robust gene knock-down.
In order not to repeat past mistakes, it is imperative to proceed with caution and use multiple methods to establish gene function.
Want to receive regular blog updates? Sign up for our siTOOLs Newsletter:
References:
Barbaric, I., Miller, G. & Dear, T. N. Appearances can be deceiving: Phenotypes of knockout mice. Briefings Funct. Genomics Proteomics 6, 91–103 (2007).
Cerikan, B. et al. Cell-Intrinsic Adaptation Arising from Chronic Ablation of a Key Rho GTPase Regulator. Dev. Cell 39, 28–43 (2016).
Kuscu, C., Arslan, S., Singh, R., Thorpe, J. & Adli, M. Genome-wide analysis reveals characteristics of off-target sites bound by the Cas9 endonuclease. Nat Biotechnol 32, 677–683 (2014).
Hannus, M. et al. siPools: highly complex but accurately defined siRNA pools eliminate off-target effects. Nucleic Acids Res. 42, 8049–61 (2014).
Liang, X. et al. Rapid and highly efficient mammalian cell engineering via Cas9 protein transfection. J. Biotechnol. 208, 44–53 (2015).
Liu, X. et al. Sequence features associated with the cleavage efficiency of CRISPR/Cas9 system. Sci. Rep. 6, 19675 (2016).
Liu, Y. et al. Targeting cellular mRNAs translation by CRISPR-Cas9. Nat. Publ. Gr. 2–10 (2016). doi:10.1038/srep29652
Marine, S., Bahl, A., Ferrer, M. & Buehler, E. Common seed analysis to identify off-target effects in siRNA screens. J. Biomol. Screen. 17, 370–8 (2012).
Rossi, A. et al. Genetic compensation induced by deleterious mutations but not gene knockdowns. Nature 524, 230–233 (2015).
Smith, C. et al. Whole-Genome Sequencing Analysis Reveals High Specificity of CRISPR/Cas9 and TALEN-Based Genome Editing in Human iPSCs. doi:10.1016/j.stem.2014.06.011
Teng, X. et al. Genome-wide Consequences of Deleting Any Single Gene. Mol. Cell 52, 485–494 (2017).
Tsai, S. Q. et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat Biotech 33, 187–197 (2015).
Unniyampurath, U., Pilankatta, R. & Krishnan, M. N. RNA Interference in the Age of CRISPR : Will CRISPR Interfere with RNAi ? (2016). doi:10.3390/ijms17030291
Veres, A. et al. Low incidence of Off-target mutations in individual CRISPR-Cas9 and TALEN targeted human stem cell clones detected by whole-genome sequencing. Cell Stem Cell 15, 27–30 (2014).
Yang, L. et al. Targeted and genome-wide sequencing reveal single nucleotide variations impacting specificity of Cas9 in human stem cells. Nat. Commun. 5, 1–6 (2014).
Further helpful reading:
Housden, B. E. et al. Loss-of-function genetic tools for animal models: cross-species and cross-platform differences. Nat. Publ. Gr. (2016). doi:10.1038/nrg.2016.118