A journey into the gut microbial control center: small RNA’s influence on Bacteroides thetaiotaomicron’s metabolism

Bacteroides thetaiotaomicron is a commensal bacterium that inhabits primarily the human large intestine and is considered one of the most important members of this microbial community. B. thetaiotaomicron is a highly versatile microbe, capable of utilizing a wide range of carbohydrates including those that are indigestible by human enzymes. It breaks down complex polysaccharides from plant cell walls and other dietary sources, producing short-chain fatty acids (SCFAs) that are an important energy source for humans. Furthermore, it has also been shown to play a crucial role in our immune system development, through the production of regulatory T-cells that help prevent autoimmune disorders.
It’s no wonder Bacteroides thetaiotaomicron has been chosen as a model representative of the gut microbiota. B. thetaiotaomicron is widely spread in human populations and relatively easy to grow and study under laboratory conditions. In a study by Ryan et al. (2020), differential RNA sequencing (dRNA-Seq) was used to generate a single-nucleotide resolution transcriptome map of B. thetaiotaomicron. High-resolution RNA-sequencing served as a tool to explore the role of small RNA molecules in regulating metabolism in the gut bacterium Bacteroides thetaiotaomicron.
By comparing different laboratory growth conditions, the researchers identified various small RNA molecules that exhibited differential expression patterns. These small RNAs were found to be associated with the regulation of key metabolic pathways in the bacterium. The authors suggest that these findings could have implications for understanding the interactions between gut microbes and the host and for the development of new therapies for metabolic diseases. Overall, the study highlights the importance of transcriptome mapping in uncovering novel regulatory mechanisms in bacterial metabolism. Furthermore, the results shed light on the intricate regulatory networks within this gut microbe and provide insights into its adaptation to different nutrient environments.
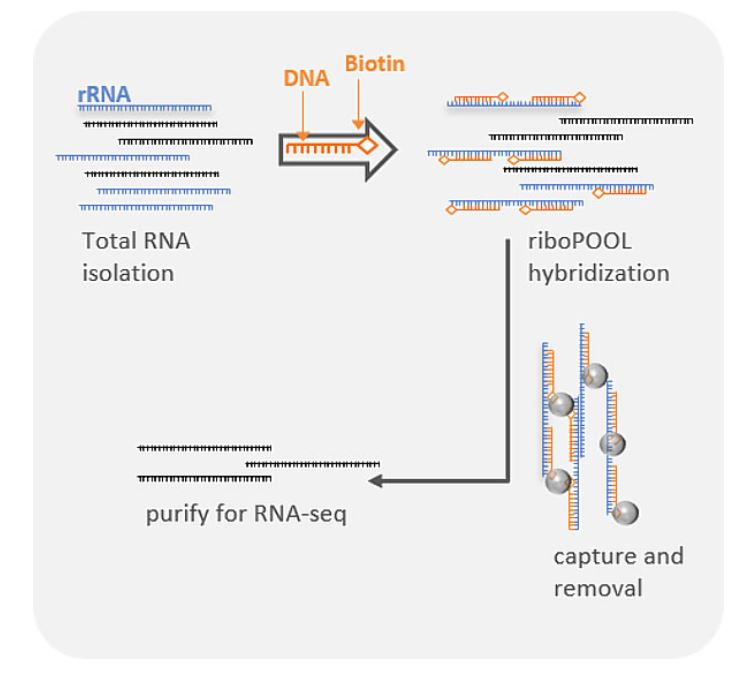
Note: Our Pan-Prokaryote riboPOOL played a small but significant role in this study. Prior to RNA sequencing, the Pan-Prokaryote riboPOOLs kit was used for ribosomal RNA depletion. Our Pan-riboPOOLs are a versatile solution that allows for simple mono- and multitranscriptomic studies using a single-step rRNA depletion for a phylogenetic group (e.g., bacteria, fungi, or mammals).
A Brief Interview with Dr. Daniel Ryan

We interviewed the first author of the study: “A high-resolution transcriptome map identifies small RNA regulation of metabolism in the gut microbe Bacteroides thetaiotaomicron”. Dr. Daniel Ryan is a postdoc at the Helmholtz Institute for RNA-based Infection Research located in Würzburg, Germany. He is a member of the Westermann Lab and his research focuses on non-coding RNAs and RNA-binding proteins in the human gut commensal Bacteroides thetaiotaomicron.
He further explained the process of generating a high-resolution transcriptome for Bacteroides thetaiotaomicron and the exciting parts of being an RNA research scientist.
- Can you briefly explain the main findings of your research and what motivated you to study small RNA regulation in Bacteroides thetaiotaomicron?
The gut microbiota has recently attracted significant attention from the scientific community due to its impact on human health and physiology. Various diseases, including inflammatory bowel disease (IBD), diabetes, colon cancer, and depression, have been linked to an imbalanced microbiota, also known as dysbiosis. Furthermore, a healthy gut microbiota plays a crucial role in preventing invasive pathogens from gaining a foothold and establishing infections. My research at the Westermann Lab aims to understand the diverse interactions between gut microbes and their host. To achieve this goal, we utilize the gut model organism Bacteroides thetaiotaomicron (B. theta), an anaerobic, non-spore forming predominant member of the healthy gut microbiota.
My foray into small RNA biology started during my Master’s thesis work at the KU Leuven, Belgium where I investigated the regulatory networks of sRNAs in E. coli. I came to appreciate the immense regulatory potential of these non-coding molecules in governing rapid responses to diverse environmental cues. A few years later, during my PhD research at KIIT University, India, I had the opportunity to delve into the roles of sRNAs in regulating acid stress survival and virulence programs in Salmonella, the pathogen responsible for causing typhoid. Having gained extensive experience in studying sRNAs and their intricate regulatory networks, I shifted gears to the gut microbiota as the focus of my postdoctoral research. After more than a decade of involvement in the field of sRNAs, I remain highly enthusiastic about uncovering novel sRNAs and investigating their intricate interactions within diverse organisms. Ultimately, my aim is to reveal regulatory cascades and pathways that can be harnessed to improve human health.
- What tools were necessary to create a high-resolution transcriptome map for Bacteroides thetaiotaomicron, and what challenges did you encounter during this process?
In order to construct a high-resolution transcriptome map of B. theta, it is crucial to extract RNA of high quality from representative and diverse conditions that effectively stimulate gene expression. This is easier said than done, since one of the main challenges of working with gut microbes is their anaerobic nature and often cumbersome culture conditions. To ensure optimal anaerobic conditions, all media and equipment used for bacterial culture must be degassed to remove oxygen, which can be toxic and inhibit growth. Once robust and reproducible growth can be achieved, RNA is extracted, sequenced and analyzed to obtain a single nucleotide resolution of the transcriptome. I then employed a suite of bio-informatics tools to annotate the transcriptome and subsequently manually validate and edit each feature. Although this final step is time-consuming and labor-intensive, it is essential for obtaining a high-quality and reliable result.
- Were there any unexpected or surprising findings in your research?
I was delighted to discover that the number of potential sRNAs in B. theta was similar to that of other model organisms, such as E. coli and Salmonella. Moreover, the vast majority of these sRNAs had no known homologs in these well-known species. This suggests that B. theta has undergone functional adaptations specific to its niche, which is primarily the human large intestine. Consequently, I anticipate a wealth of novel biological insights, potentially revealing new modes of interaction and target regulation.
- Are there any potential applications or implications of your research for human health, such as developing targeted therapies or interventions for gut-related disorders?
In order to develop effective targeted therapies, it is crucial to first and foremost “know your target”. Going in blind is never a good strategy and this is where I see the potential of this work. With this high-resolution transcriptome and the “Theta-Base” browser, we have provided a framework to discover and identify novel genes and sRNAs that can further be investigated as potential targets to regulate or modulate activity. These newly identified targets whether coding or non-coding can be exploited to modulate B. theta to achieve specific functions for instance, they could be used to exclusively metabolize a particular carbon source or prevent the consumption of a specific metabolite. While this example is simplistic, several laboratories are already conducting pilot studies in this area, offering promise for the future of targeted medicine.
- What would you say are some of the challenges or gaps in knowledge that need to be addressed in the field of gut microbiota research?
One of the overarching challenges in gut microbiota research is distinguishing between correlative and causative effects. It is therefore imperative to develop protocols and methodologies that delve into various phenomena at a detailed level before drawing reliable conclusions.
There are also specific challenges to address, particularly regarding the creation of microbial consortia that accurately reflect the composition of the gut microbiota. This is not easy considering the vast numbers of bacteria and their complex interactomes that make up a healthy microbiota. Moreover, models representing the human intestinal niche, which harbor these diverse microbial communities, need further refinement to better reflect this complex environment.
- Finally, what is your favorite part of being an RNA research scientist?
As an RNA research scientist, what I find most fascinating is the varied range of roles that this molecule can play. From intricate structural scaffolds to subtle enzymatic and regulatory functions, RNA displays a multitude of capabilities, and witnessing these firsthand is truly captivating.
Biocabulary
Differential RNA sequencing (dRNA-Seq) is a technique used to identify transcriptome features and define overall transcriptomic architecture, such as transcription start sites, terminators, non-coding RNAs, coding RNAs, promoters, etc.
A transcriptome map is a comprehensive profile or catalog of all the RNA molecules (transcripts) produced by an organism or a specific cell type under particular conditions. It provides a snapshot of the active genes and their expression levels within the cells or tissues being studied.









{kind=link}