Disrupting lncRNA function with siPOOLs (RNAi), antisense oligos and CRISPR
Summary
This blogpost covers methods used in the disruption of lncRNA function. Specifically focusing on RNA interference (with siPOOLs), antisense oligos, and CRISPR approaches. Challenges faced with these approaches are addressed.
Long non-coding RNAs (lncRNAs) make up a major subgroup of RNAs and are defined as over 200 nucleotides long with limited protein-coding potential. There are three times as many genes producing lncRNAs as opposed to proteins. Numerous studies have described functional roles of lncRNAs in development and disease. This has stimulated major global interest and intense efforts to decode lncRNA function.
Disrupting lncRNA function
One way to find out what a lncRNA does is to decrease its expression, thereby disrupting its function. Current methods of downregulating lncRNA expression include knockdown approaches with siRNA and antisense oligos (ASOs), or knockout approaches with CRISPR, TALENs and other techniques involving DNA nucleases.
As we have mentioned before, knockdown and knockout approaches employ different mechanisms and as a result sometimes yield different results. Hence it is highly recommended to employ both techniques when possible to thoroughly validate lncRNA function.
LncRNA functional knockdown – RNAi and antisense approaches
LncRNA knockdown involves the transient downregulation of lncRNAs at the RNA level. This typically involves RNA degradation mediated by the RNA interference (RNAi) machinery for siRNAs, or with RNase H for ASOs.
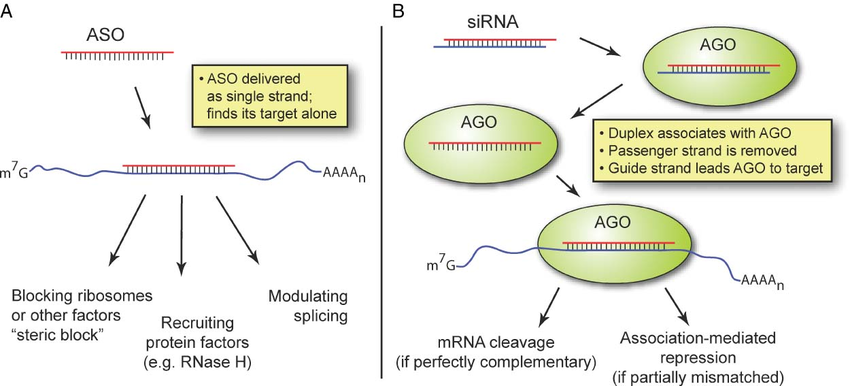
Figure from Watts, J. K. & Corey, D. R. Silencing disease genes in the laboratory and the clinic. J. Pathol. 226, 365–79 (2012).
Some challenges that both technologies face when targeting lncRNAs:
- low endogenous expression of lncRNA may limit efficiency of knockdown
- accessibility of siRNA/ASO to lncRNA may be limited by secondary structure (created by folding of the lncRNA and self-base pairing)
- accessibility to siRNA/ASO to lncRNA may be limited by bound proteins
- off-target effects
Does cellular localization matter when disrupting lncRNA function?
Cellular localization of lncRNAs was reported to account for differences in knockdown efficiency by ASOs compared with siRNAs. Although there have been observations that RNAi factors are present in the nuclei, siRNAs were reportedley less efficient than ASOs for modulating nuclear-localized lncRNAs (Lennox and Behlke, Nucleic Acids Res, 2016).
This does not appear to apply to all cases as using siPOOLs (high complexity pooled siRNA) or ASOs led to similar downregulation of NEAT1, a nucleus-localized lncRNA:
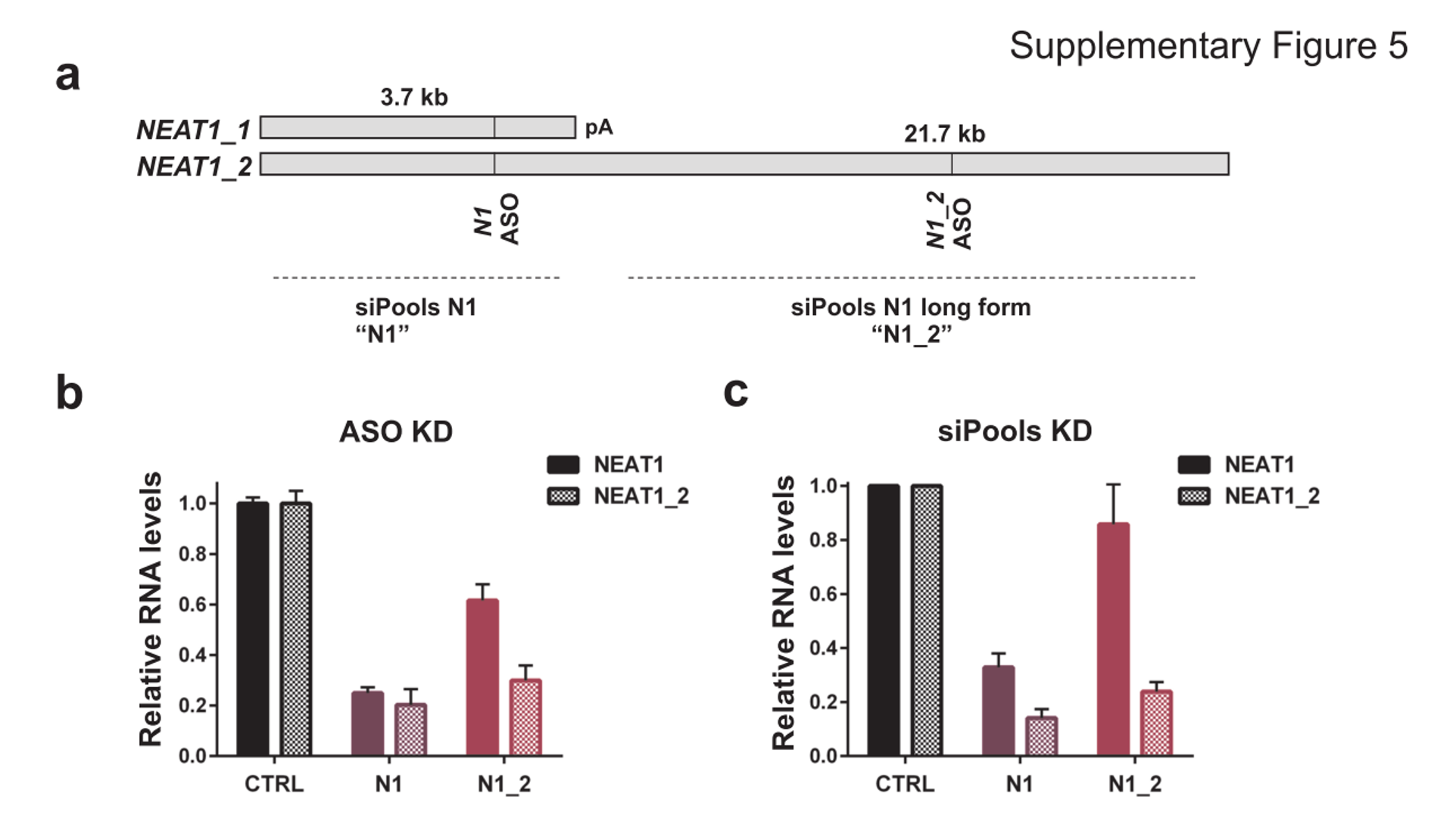
NEAT1 lncRNA has two isoforms, 3.7kb NEAT1_1 and longer 21.7kb NEAT1_2. MCF7 cells were transfected with either LNA GapmeRs (ASOs) or siPOOLs that target both isoforms (N1) or the long form only (N1_2). RNA levels of both isoforms (NEAT1) or only the long isoform (NEAT1_2) were quantified after 24h. (Adriaens et al., Nat Med, 2016)
siPOOLs also worked well for XIST and MALAT1 (~80% KD at 1 nM), both nuclear-localized lncRNA. Notably however, cytosolic-localized lncRNAs such as H19 were much more efficiently targeted with the high complexity siRNA pools (> 95% KD at 1 nM).
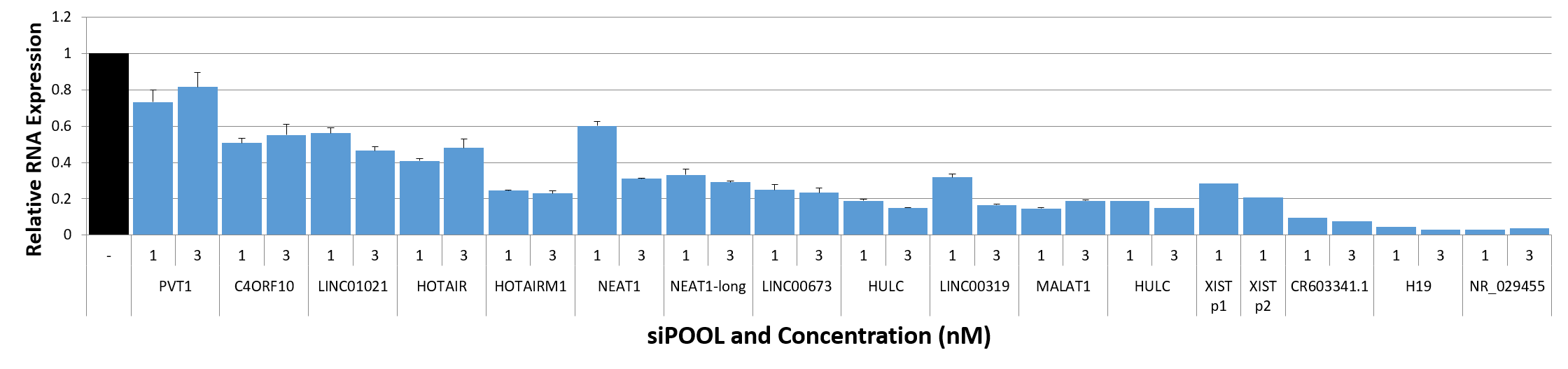
siTOOLs Biotech in-house data showing knockdown efficiencies of siPOOLs against 16 lncRNAs tested at 1 or 3 nM in standard cell lines (MCF7, A549, Huh7). Assayed by real-time quantitative PCR after 24h.
Compared to coding genes, the above-mentioned factors do limit efficiencies of knockdown approaches. But with siPOOLs, the greater diversity of siRNA sequences is expected to increase chances of association with the target RNA. In-house data shows 12 of 16 tested lncRNAs showed good knockdown efficiencies of > 70% with siPOOLs.
Importantly, siPOOLs efficiently counter off-target effects commonly associated with siRNA. Off-target effects have also been reported to occur with ASOs, especially since they are also exposed to intronic regions. Hepatotoxicity related to certain sequence motifs on LNA-modified ASOs have also been reported (Burdick et al., 2014)
lncRNA functional knockout with CRISPR
The genomic distribution of lncRNA loci is rather complex. They are typically categorized in relation to their proximity with protein coding genes.
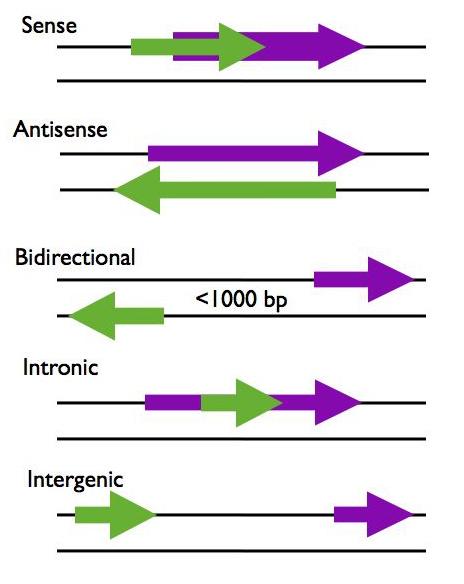
Figure showing lncRNA loci in green and protein-coding loci in purple. Arrows indicate direction of transcription. Figure and description below from McManus lncRNA presentation: https://mcmanuslab.ucsf.edu/node/251
- Sense – The lncRNA sequence overlaps with the sense strand of a protein coding gene.
- Antisense – The lncRNA sequence overlaps with the antisense strand of a protein coding gene.
- Bidirectional – The lncRNA sequence is located on the opposite strand from a protein coding gene whose transcription is initiated less than 1000 base pairs away.
- Intronic – The lncRNA sequence is derived entirely from within an intron of another transcript. This may be either a true independent transcript or a product of pre-mRNA processing
- Intergenic – The lncRNA sequence is not located near any other protein coding loci.
Hence disrupting lncRNAs with DNA nucleases can be a challenging affair that runs the risk of affecting neighbouring genes.
How many lncRNAs can be CRISPRed?
Goyal et al. 2017 performed a genome-wide “CRISPRability” analysis to evaluate the risks and utility of CRISPR for disrupting lncRNA function.
Introducing mutations with CRISPR is generally not applicable for lncRNAs. Mainly due to difficulty predicting active functional domains and the fact that some lncRNAs exert phenotypes through the act of transcription per se.
Deleting the entire lncRNA is an option but not when it overlaps with other genes. Hence, the major approach is to target lncRNA promoters. But then we run into the problem of affecting neighbouring genes that share promoters.
So they came up with three “CRISPRability” rules to avoid potential effects on neighbouring genes:
Rule 1: Sense, antisense and intergenic lncRNAs are considered “non-CRISPRable” when transcribed from bidirectional promoters, defined by presence of another promoter present 2000bp upstream/downstream of lncRNA start.
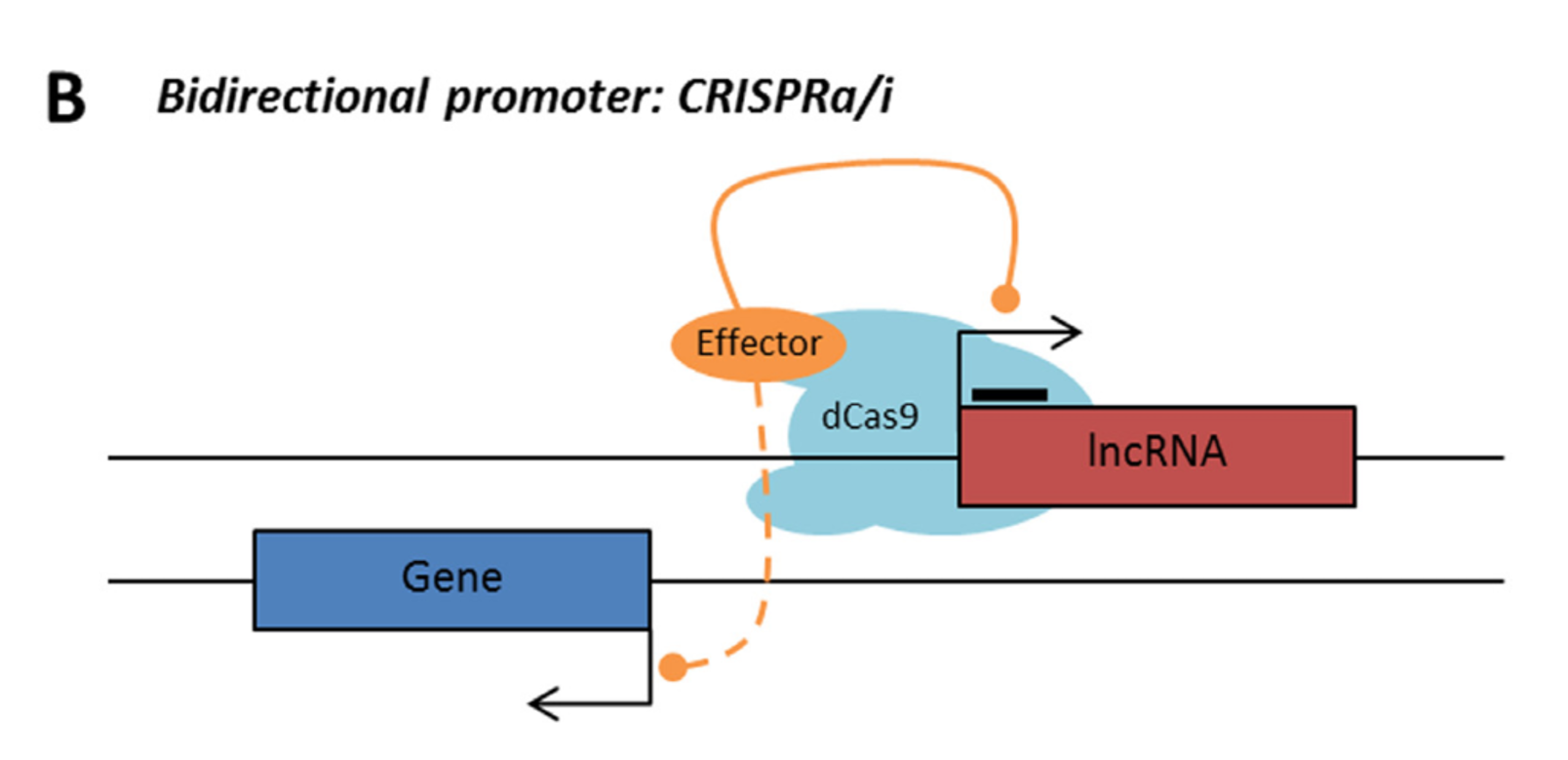
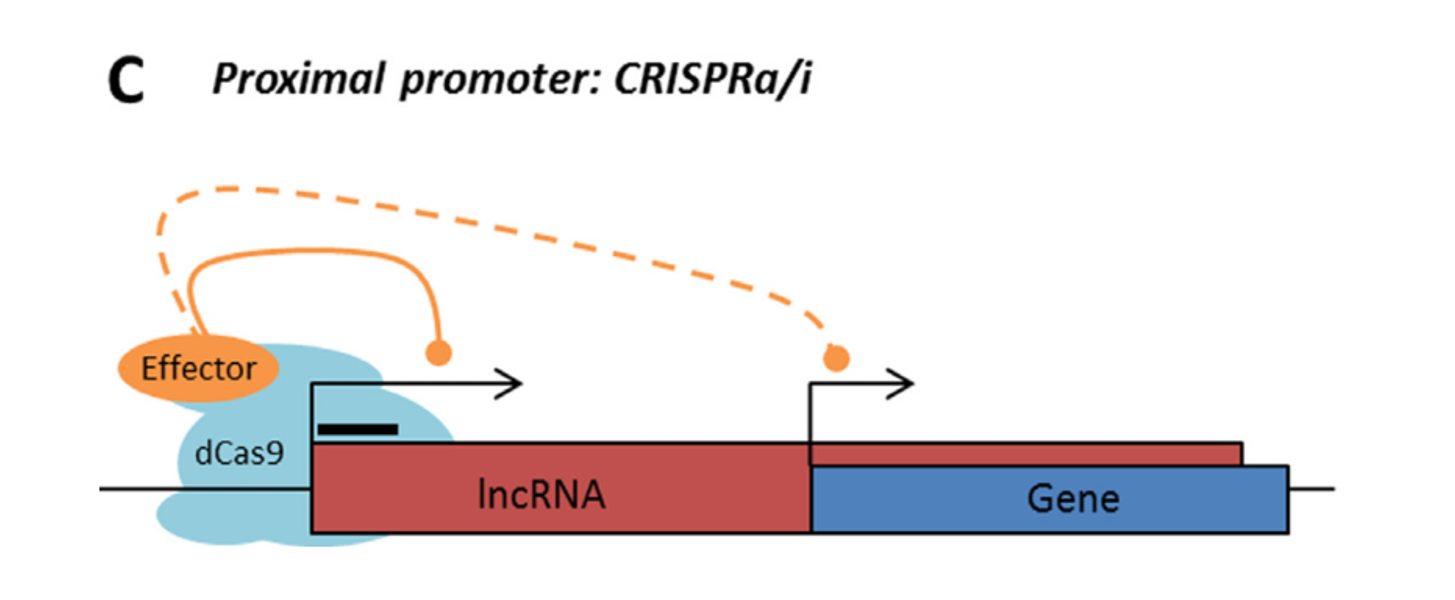
Rule 2: Sense, antisense and intergenic lncRNAs are considered “non-CRISPRable” when the start of the lncRNA is located closer than 2000p to the start of the neighbouring gene, excluding lncRNAs transcribed from bidirectional promoters – termed “proximal promoters“.
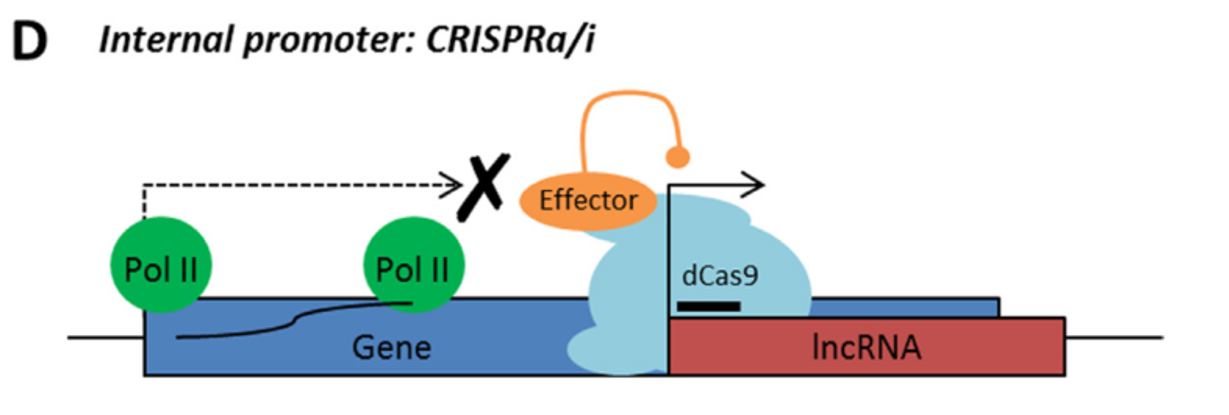
Rule 3: Sense and antisense lncRNAs are considered “non-CRISPRable” when transcribed from internal promoters, where the start of the lncRNA falls within the gene body of another coding/non-coding transcript. This would include intronic lncRNAs.
After applying “CRISPRability” rules, only 38% of all lncRNAs were suitable for CRISPR-based functional disruption

Figure from Goyal et al., 2017 showing proportion of lncRNAs that fall within the 3 rules of “CRISPRability”
The study went on to corroborate the relevance of the classification by testing effects of CRISPR/Cas9 compared to ASOs/siRNA on their targets and neighbouring genes.

An example involved lncRNA HOTAIR that arises from the HOXC locus which regulates expression of several genes including HOXC11. They found that dCas9-KRAB , which produces CRISPR-based transient inhibition (CRISPRi) by blocking transcription, caused knockdown of HOXC11 when designed to target HOTAIR. This occurred for all 3 independent sgRNAs. siPOOL-mediated knockdown of HOTAIR, in contrast, did not affect HOXC11.
Similar scenarios were seen with coding genes, in particular for well-known tumour suppressor TP53, where neighbouring gene WRAP53alpha tended to be downregulated by dCas9-KRAB. This effect was absent with siPOOLs targeting TP53.
It therefore pays to carefully note the genomic neighbourhood of lncRNAs when using CRISPR for disruption. A careful scientist would also monitor the expression of neighbouring/overlapping genes in parallel to the target gene. Orthogonal methods such as RNAi (with siPOOLs), or rescue experiments that restore expression of the lncRNA, is recommended to fully evaluate lncRNA function.
Learn more about siPOOLs!
Featured blog image from lncRNA blog, photo credit autism.am