Low complexity pooling does not prevent siRNA off-targets
Summary: Low-complexity siRNA pooling (e.g. Dharmacon siGENOME SMARTpools) does not prevent siRNA off-targets. It may in fact exacerbate off-target effects. Only high-complexity pooling (siPOOLs) can reliably ensure on-target phenotypes.
Low-complexity pooling increases the number of siRNA off-targets
One of the claims often made in favour of low-complexity pooling (e.g Dharmacon siGENOME SMARTpools) is that this pooling reduces the number of seed-based off-target effects compared to single siRNAs.
If this were true, we would expect different low-complexity siRNA pools for the same gene to give similar phenotypes. But this is not the case.
Published expression data shows that low-complexity pooling actually increases the number of off-targets.
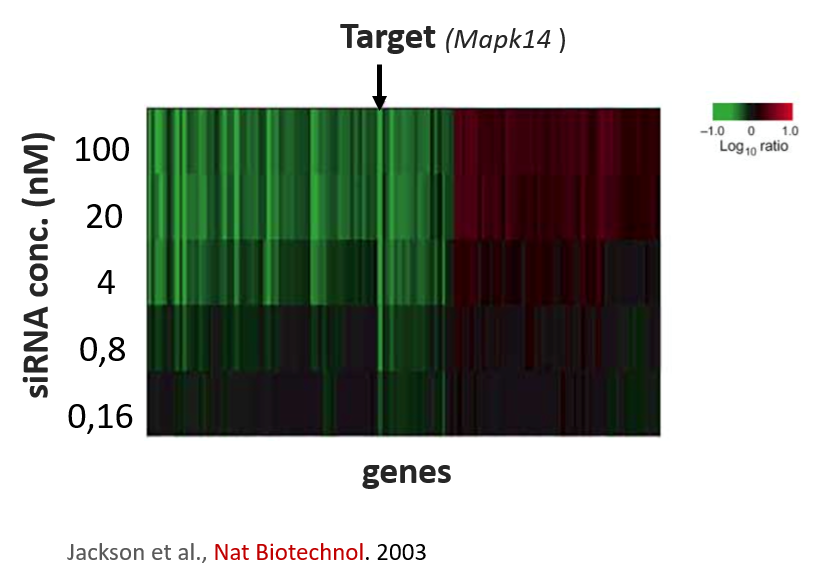
Kittler et al. (2007) looked at the effect of combining differing number of siRNAs in low to medium complexity siRNA pools (siRNA pools sizes were: 1, 3, 5, 9, and 12).
Their work showed that the number of down-regulated genes (50% or greater silencing) actually increases when small numbers of siRNAs are combined. Only when larger numbers of siRNAs are combined does the number of off-targets start to drop:
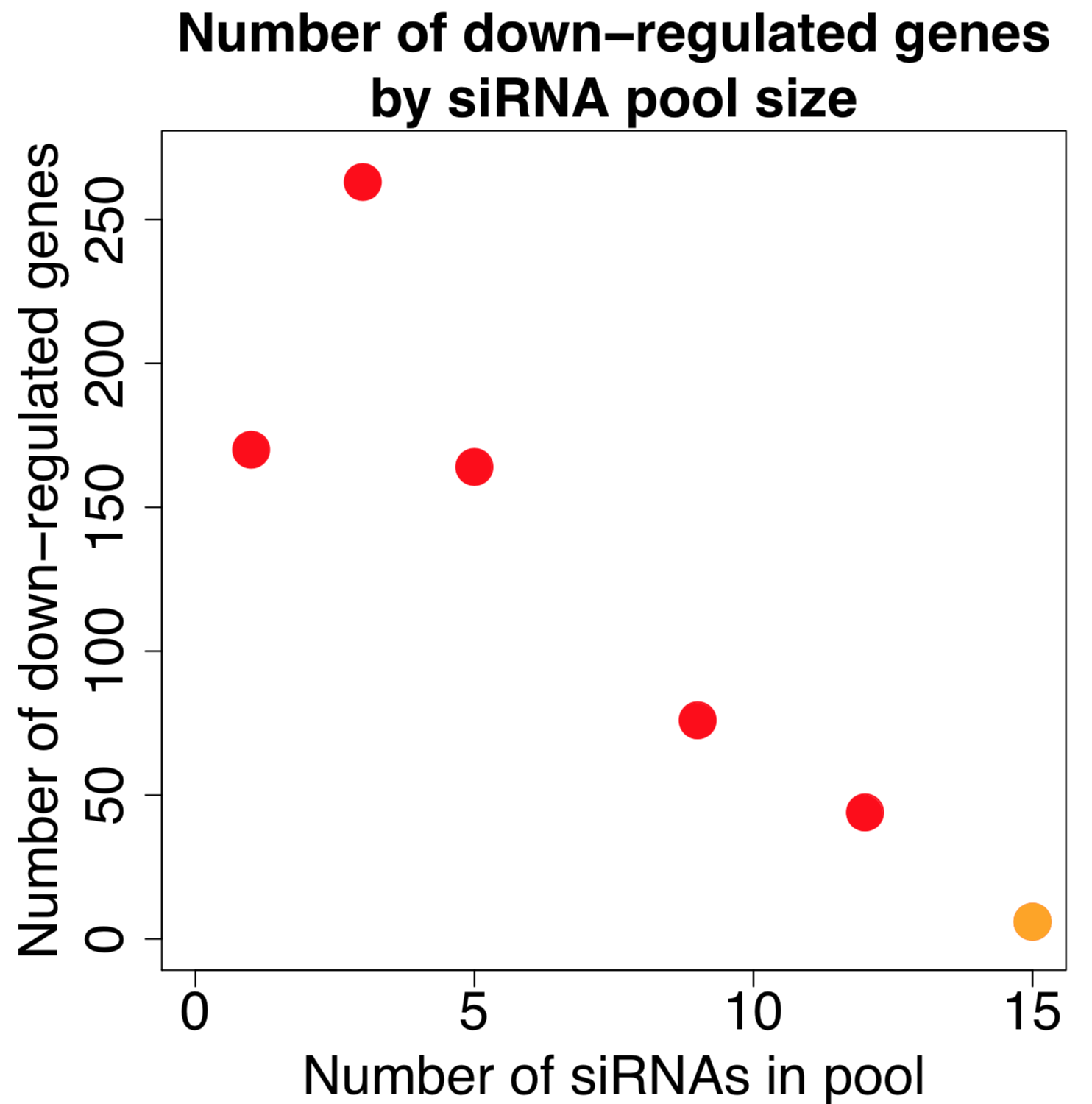
[The figure is based on data from GEO dataset GSE6807. Down-regulated genes are those whose expression is reduced by 50% or more. Note that the orange point is taken from our 2014 NAR paper, as we are not aware of other published expression datasets with this many pooled siRNAs. A few caveats with combining these datasets are that they use different target genes, siRNA concentrations, and the data comes from a different expression platform.]
Low-complexity pooling: a bad solution for siRNA off-targets
Low-complexity pooling does not get rid of the main problem associated with single siRNAs: seed-based off-target effects. Based the above analysis, it can make it even worse. It also prevents use of the most effective computational measures against seed effects.
Redundant siRNA Activity (RSA) is a common on-target hit analysis method for single-siRNA screens. It checks how over-represented the siRNAs for a gene are at the top of a ranked screening list. If a gene has 2 or more siRNAs near the top of the list, it will score better than a gene that only has a single siRNA near the top of the list. This is one way to reduce the influence of strong off-target siRNAs.
Correcting single siRNA values by seed medians has also been shown to be an effective way to increase the on-target signal in screens. This correction is not effective for low-complexity pools, since each pool can contain 3-4 different seeds.
Off-target based hit detection algorithms (e.g. Haystack and GESS) are also only effective for single-siRNA screens. The advantage of these algorithms is that it permits the detection of hit genes that were not screened with on-target siRNAs. These algorithms are not effective for low-complexity pool screens.
Our recommendation: do not convert single siRNAs into low-complexity pools, rather use high-complexity siPOOLs to confirm hits
We do not recommend that screeners combine their single siRNA libraries into low-complexity pools (e.g. combining 3 Silencer Select siRNAs for the same target gene). If possible, it is better to screen the siRNAs individually and then apply seed-based correction, RSA and seed-based hit-detection algorithms.
The time saved by only screening one well per target may prove illusory when the deconvolution experiments show that the individual siRNAs have divergent phenotypes.
It is probably better to deal with off-target effects up front (by screening single siRNAs) than to be surprised by them later in the screen (during pool deconvolution).
Reliable high-complexity siPOOLs, as independent on-target reagents, can then be used to confirm screening hits.
siTOOLs also now has RNAi screening libraries available. Please contact us for more information.