5 factors to consider in multi-gene targeting RNAi screens
Summary: Effective functional genomic screening depends on a variety of factors that need to be simultaneously addressed to obtain meaningful results. A recent Cell Reports paper demonstrates this by taking a holistic approach to siRNA screening with the use of multi-isoform/multi-gene targeting to address redundant paralogs and pathways in cancer cells.
The case for multi-gene targeting
Many RNAi screens use arrayed single gene knockdowns to find genes that play an important role in a biological process. The idea is that a single bullet is enough to take down its target leaving a gaping hole that one cannot fail to notice. In some cases, this is true, and is certainly relied upon by drug developers seeking to create specific mono-target drugs. However, in complex diseases like cancer, cells have evolved fail-safe mechanisms to make them more resistant to external assaults. A single bullet is simply not enough.
Take for example oncogenic protein RAF or Rapidly Accelerated Fibrosarcoma, a tyrosine kinase effector that is a component of the MAPK signalling pathway (Ras-Raf-MEK-ERK). RAF has three isoforms – ARAF, BRAF and RAF1 (also called CRAF). Studies in mouse embryonic development show they all share some form of functional redundancy as knocking out two isoforms produces more severe effects than knocking out each isoform alone.
Screens that target single genes/isoforms therefore tends to bias results towards genes that have no paralogs or only have single isoforms. This was indeed the reason why classical Ras effectors were not identified in previous screens.
Factors to consider in a multi-gene targeting RNAi screen
Determining gene combinations that make sense
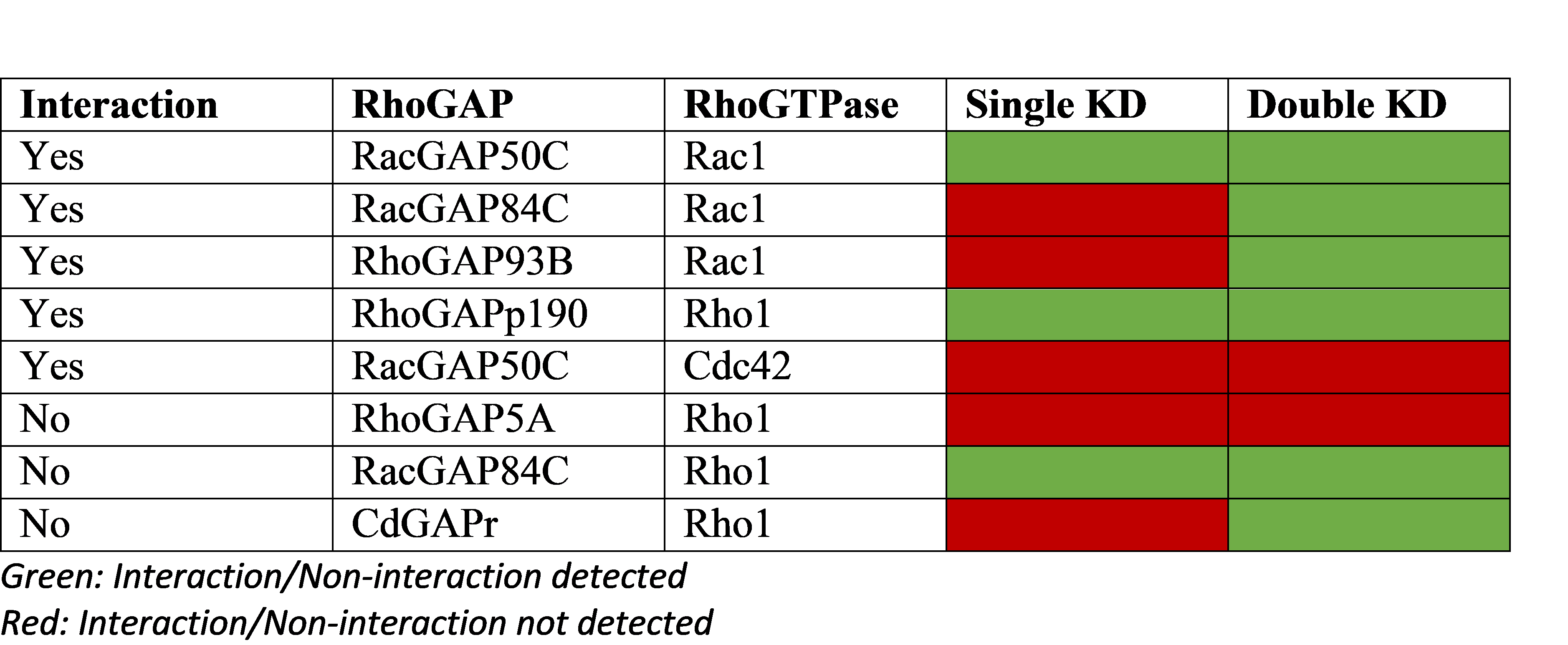
The authors of the study did a focussed siRNA screen on 41 RAS effector nodes represented by 84 genes. Out of the 41 nodes, 25 of them had 2-4 functional paralogs where combinatorial gene silencing was carried out with multiple siRNAs. 5 nodes knocked down multiple members of a protein complex. 5 nodes had siRNAs targeting multiple steps within a pathway. Only 6 nodes silenced single genes (highlighted).
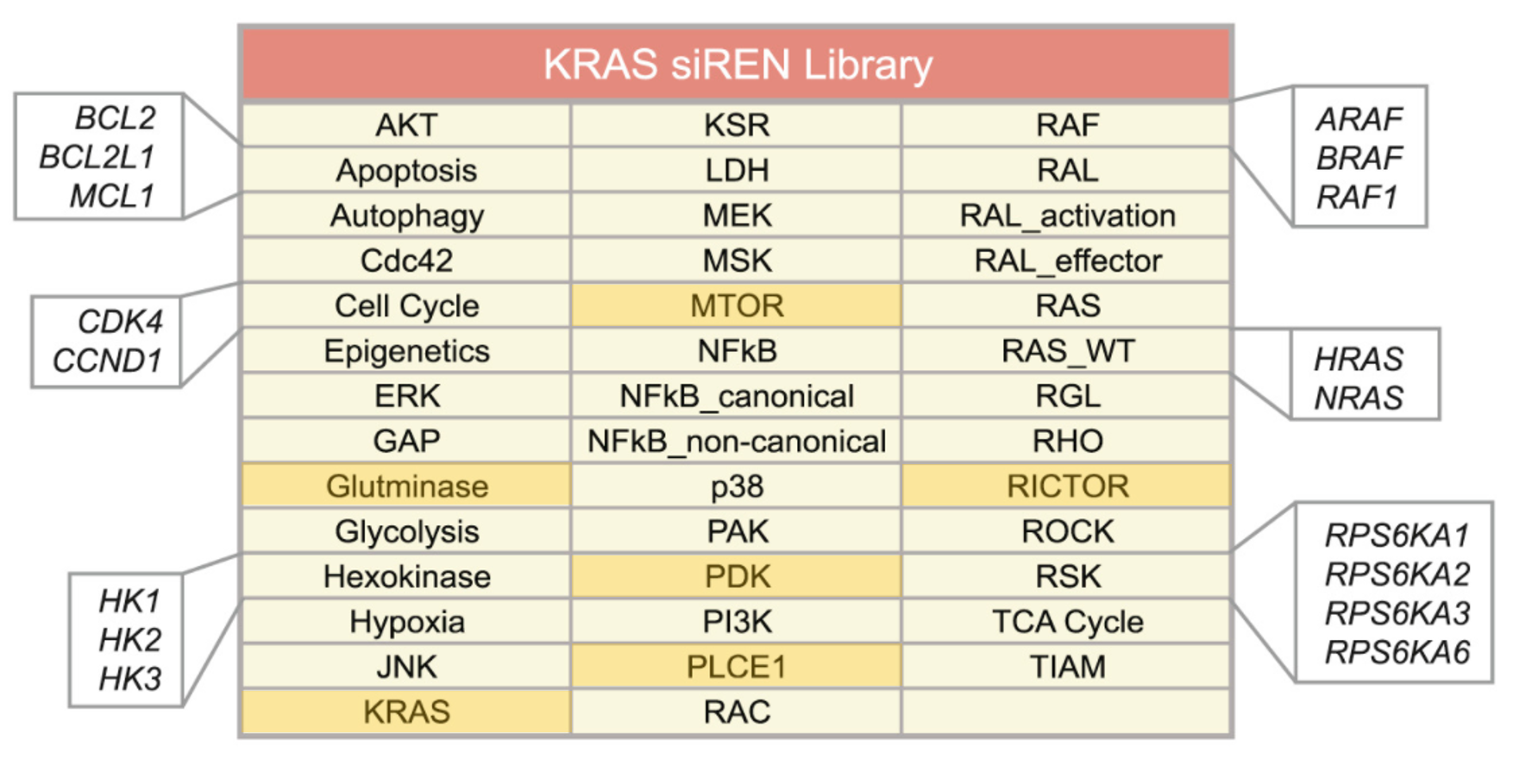
The only caveat with designing such a screen is the requirement for prior knowledge to perform meaningful gene silencing combinations. In this instance, many of the Ras effector pathways are characterized sufficiently to do this well however in other less studied fields, this could be a challenge. Useful tools that would help in designing gene knockdown combinations would include pathway or phenotype databases such as KEGG, REACTOME or Wikipathways. The Phenovault which siTOOLs Biotech is developing, is yet another potentially useful tool.. more details to come!
Number and types of phenotypes
The authors also highlight how a screen that reads only one phenotype might miss other important gene functions. Many RNAi screens sadly still stick to measuring cell proliferation as their only read-out which is greatly influenced by siRNA off-target effects. Here, 5 different phenotypes were measured (cell size, proliferation, apoptosis, reactive oxygen species [ROS], and viability). It was noted that silencing of Cdc42 had little effect on cell viability yet a prominent effect on ROS levels.
To take this up a notch, analysis was also performed at the single-cell level in cells expressing uniform levels of GFP and co-transfected with GFP siRNA. This allowed authors to correlate phenotypes with levels of gene knockdown, generating dose-response curves. How clever!
A lot more work, but adds to data robustness especially when using single siRNAs that are known to be rather variable.
Heterogeneity of cell lines
Many reports and our own observations attest to the heterogenous response of different cell lines to the same treatment. In cancer especially, the large heterogeneity necessitates the use of multiple cell lines. Not doing so would be failing to account for the large genetic diversity observed in the clinic. The authors screened 92 cell lines derived from lung, pancreas and colorectal tissue.
Despite seeing heterogenous responses to node knockdowns, phenotypic responses could be distinguished into several groups based on effector engagement. A major group dependended on RAF through direct binding with KRAS, a second major group worked via RSK p90 S6 kinases to drive RSK-mTOR signalling. And a third minor group was dependent on RalGDS. They went on to focus on the first two major groups, naming them KRAS-type and RSK-type respectively.
Reagents – choosing siRNAs and siRNA concentrations
The authors used previously characterized siRNAs to select for more potent siRNAs. This involved an RNAi sensor reporter-based assay that required the generation of 20,000 clones. The reporter was also shRNA-based. Due to heterogeneity in Dicer-mediated cleavage of shRNA, its uncertain if knockdown potency is accurately reflected when translated to siRNAs (read about the difference between shRNAs and siRNAs).
In any case, its a lot of work to characterize all siRNAs to be used in a screen. Furthermore, off-target effects are not addressed.
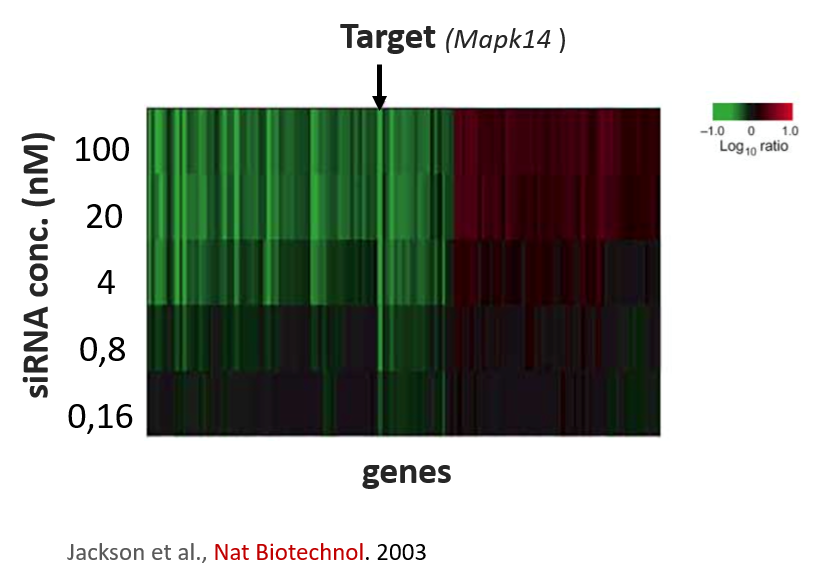
The authors stuck to a maximal concentration of 12 nM where 2 nM of siRNA was applied per gene. At 2 nM per siRNA, one still risks deregulating other genes. One of the first papers by Aimee Jackson et al., demonstrated an siRNA targeting MAPK14 deregulated many other genes even at concentrations of 1-4 nM.
An important consideration is to ensure total siRNA concentrations are kept constant. In which case, a negative control siRNA has to match or follow the maximal siRNA concentration used. Using different levels of siRNAs runs the risk of biasing off-target effects towards sequences present at higher concentrations.
To learn what the causes, extent and consequences of siRNA off-target effects are, read siTOOLs Technote 1)
Validating results
As with all scientific hypothesis, it helps to arrive at the same conclusion with different approaches.
The two different effector response subgroups identified also responded differently to small molecules. The KRAS-type lines being more sensitive to EGFR and ERK inhibition while the RSK-type lines more sensitive to inhibitors of PDK1, RSK, MTOR, S6K1 and DNA repair enzymes. This was attributed to the latter’s higher basal metabolic activity manifested in larger investments towards oxidative phosphorylation and mitochondrial ribosome maintenance.
By also projecting signatures obtained from cell lines into patient samples (in The Cancer Genome Atlas, TCGA), the subtypes were also effective at predicting differential sensitivity to multiple drug treatments. This highlights the importance in designing effective drug combinations in cancer.
Interestingly, the authors also performed CRISPR pooled screens in parallel. However, due to the restraints of being only able to knockout 1 gene at a time, smaller effects were seen due to gene redundancy. However, they did go on to use CRISPR as well to mutate key genes to affirm the pathway relationships established.
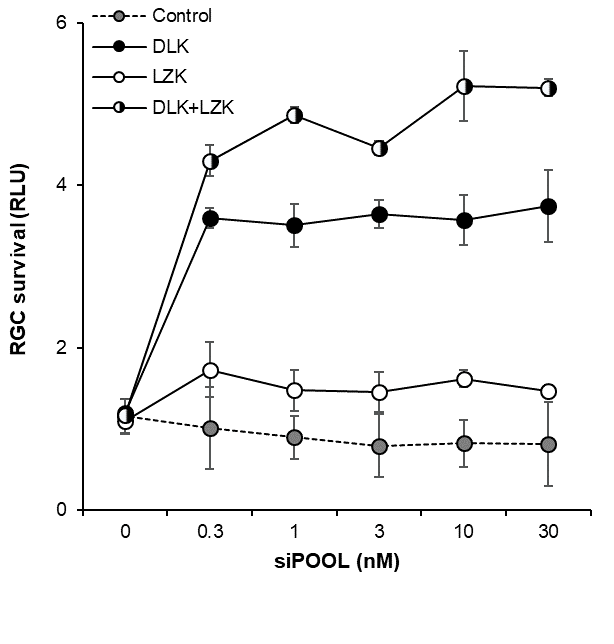
siPOOLs have been used successfully for multi-gene targeting for up to 4 genes, and potentially more. They also safely address off-target effects by high complexity pooling, enabling each siRNA to be applied at picomolar concentrations. For more articles on multi-gene targeting, read an older blogpost:
Understanding gene networks with combinatorial gene knockdown