siRNA off-target effects occur when siRNA molecules bind to and silence genes other than their intended target, leading to unintended gene expression changes. These effects can complicate results and interpretations of RNAi experiments, highlighting the need for careful siRNA design and validation strategies to minimize unintended interactions and ensure specificity in gene silencing
Although we talk a lot about off-targets, one of the main advantages of siPOOLs (complex siRNA pools) compared to single siRNAs or mini-pools (Dharmacon) is that they provide near optimum silencing of target genes. Two siPOOLs for the same gene give very similar knock down levels, and their silencing is around the best of any single siRNA. Given how many candidate siRNAs there are for a gene, and how difficult it is to accurately predict silencing levels, this makes siPOOLs the best choice for gene silencing.
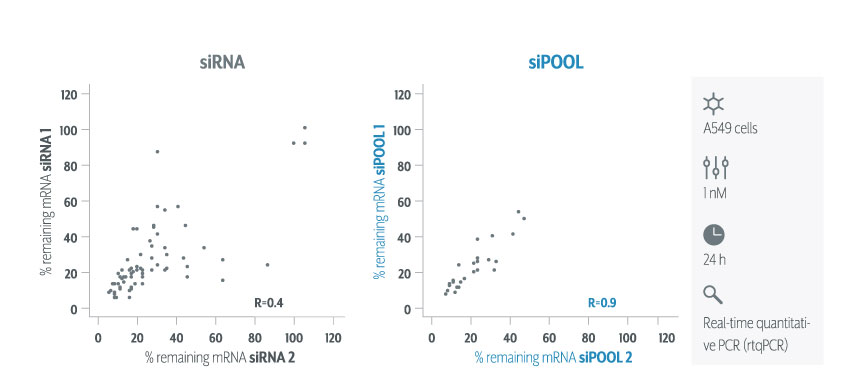
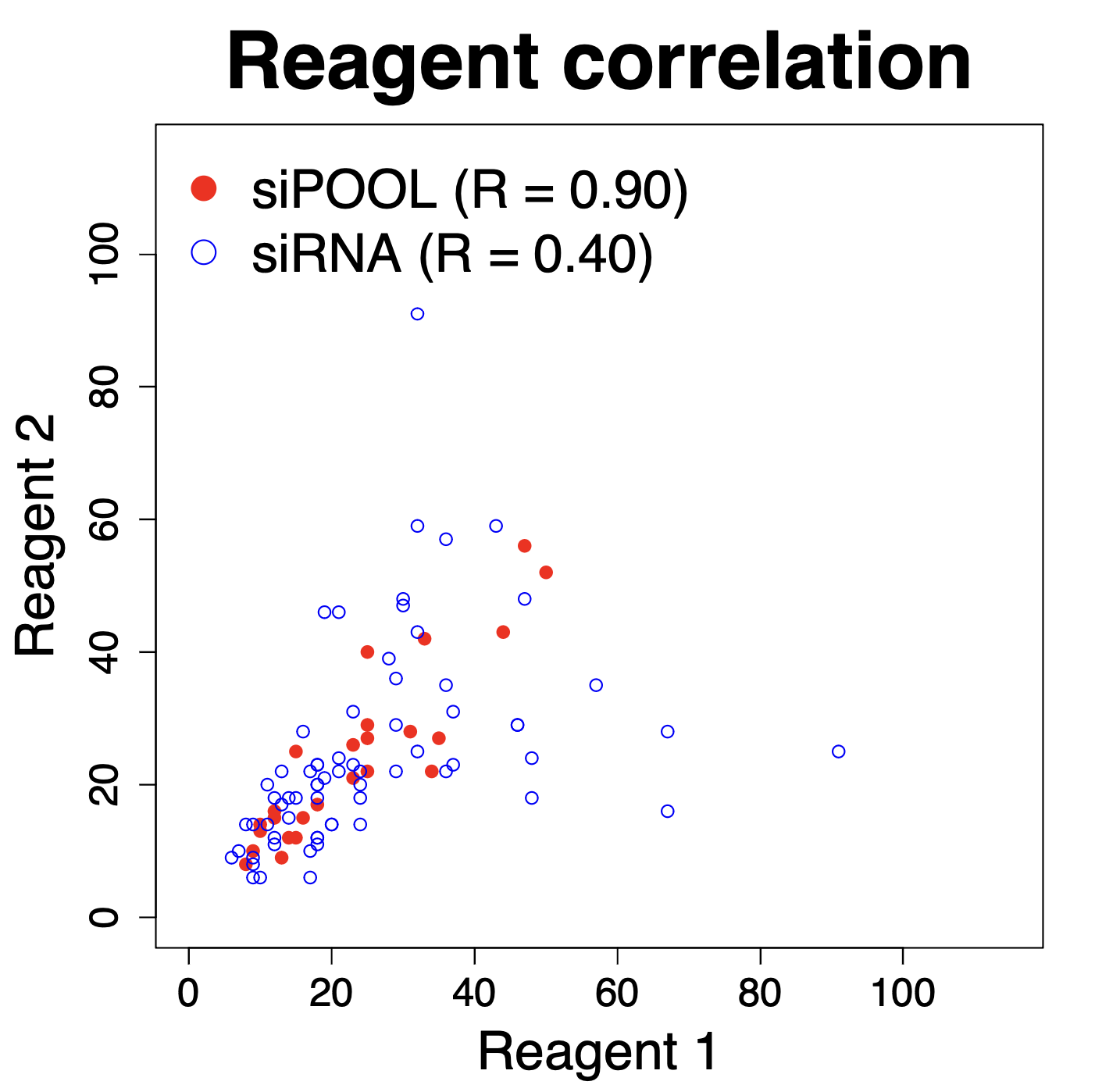
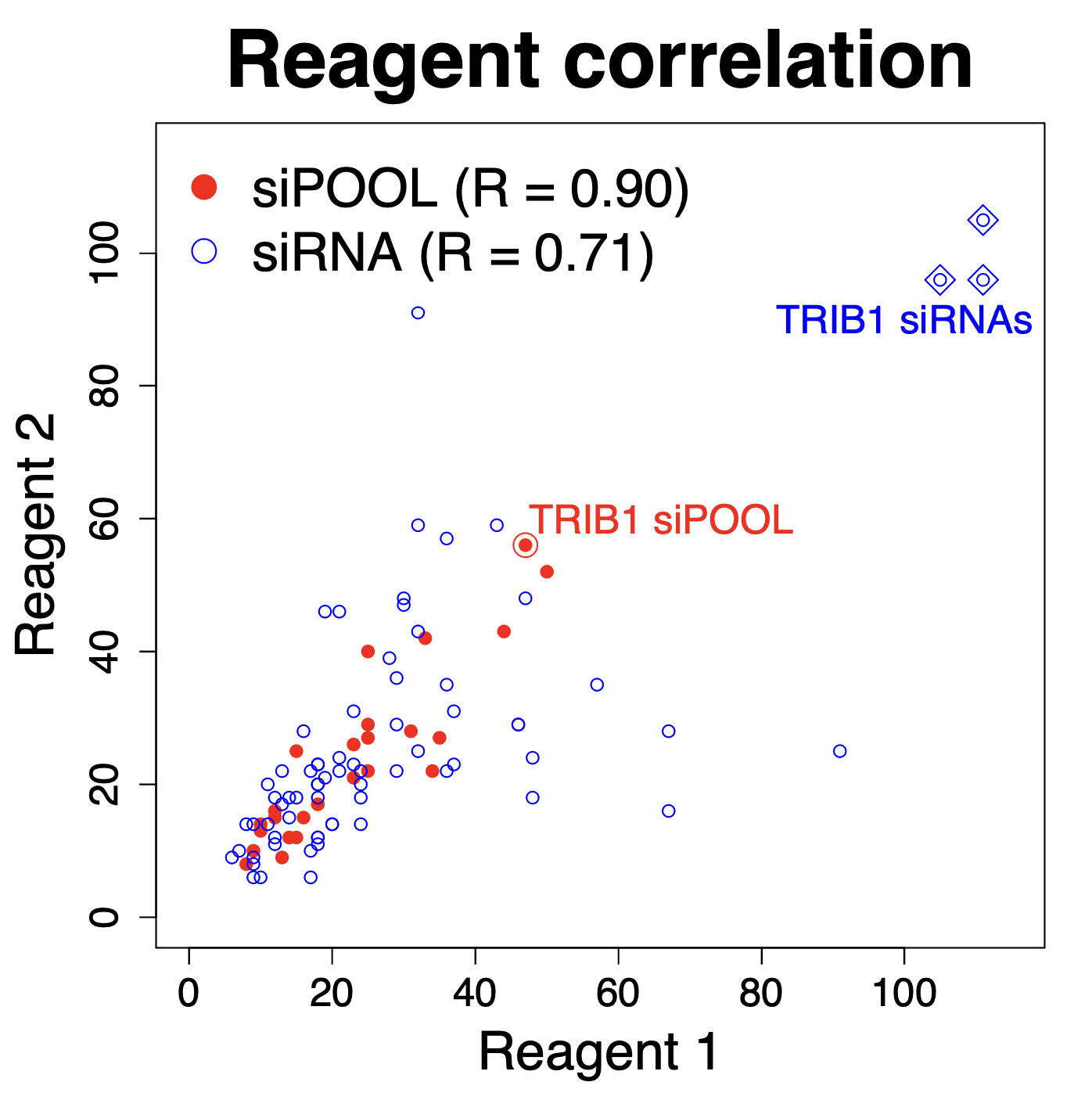
The following plot, comparing independent siPOOLs and siRNAs for the same target gene, shows that siPOOLs for the same gene give more similar silencing than do siRNAs (these are Ambion Silencer Select siRNAs).
We see that the correlation for independent siPOOLs is nearly twice that for independent siRNAs.
(Note that for siRNAs we are doing all pairwise comparisons for 3 siRNAs per target gene. Randomly selecting 2 siRNAs per gene gives similar R values.)
In the above plot, we removed 3 siRNAs that did not work, for the gene TRIB1. TRIB1 has some association with the nucleus and has a short mRNA half life, both of which are factors associated with poor gene silencing.
The following plot shows the TRIB1 siPOOLs and siRNAs.
Note that including these non-functional siRNAs actually improves the reagent correlation, though not for a good reason!
We also see that independent TRIB1 siPOOLs give very similar silencing and it’s much better than for the siRNAs. In our experience, if a siPOOL does not work well for a gene, designing a second siPOOL does not substantially improve things, as the poor silencing is normally a feature of the target gene itself. ~50% silencing is probably about the best one can expect for this gene.
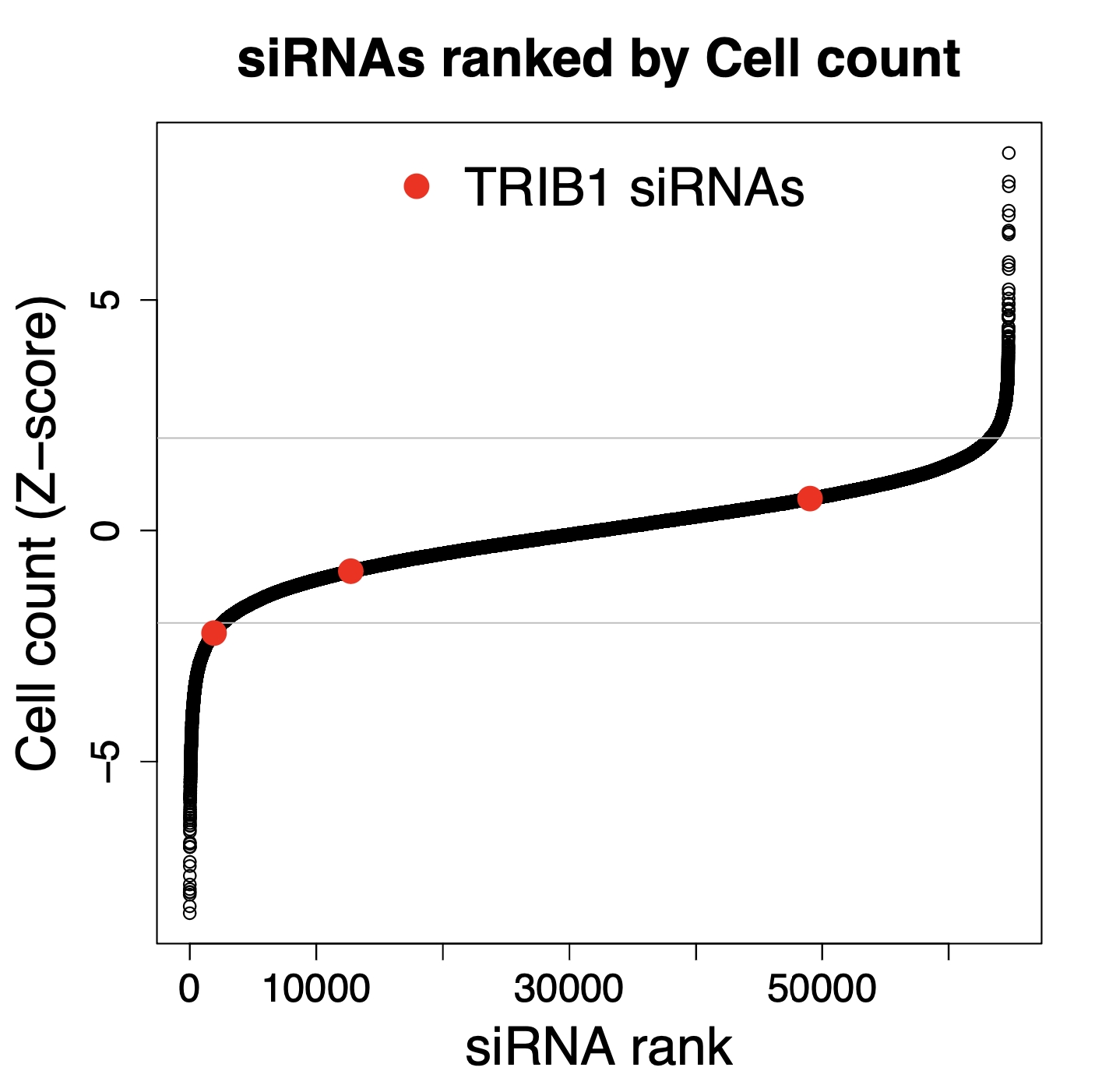
Just because siRNAs do not give any on-target silencing, this does not mean they can’t show up as hits in screening assays. Because most of the downregulation is in off-target genes (due to the seed effect), each of those TRIB1 siRNAs may silence nearly 100 genes.
We looked at a genome-wide RNAi screen that included these 3 Silencer Select siRNAs. We see that one of them gives a fairly strong phenotype (Z-score < -2 for cell count), even though the siRNAs do not silence their on-target gene.
Screening with siPOOLs is the smarter alternative, as you can be confident that they provide near optimal on-target silencing and have less off-target effects.
A 2013 study on Parkin translocation used genome-wide siRNA libraries from Ambion (single Silencer Select siRNAs) and Dharmacon (pools of 4 siGENOME siRNAs).
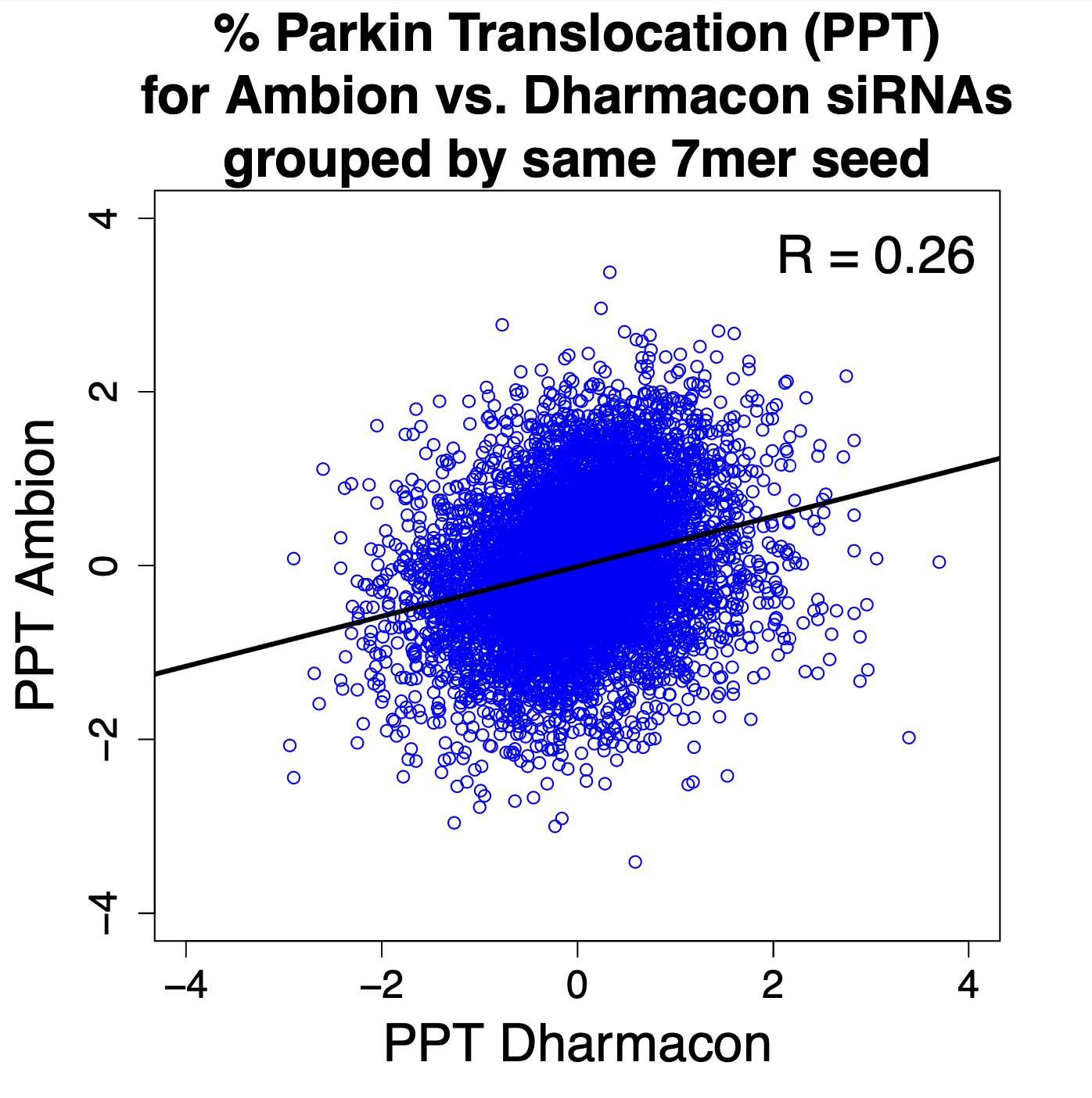
The correlation between results for the same on-target gene from the two libraries was very low (R = 0.09). (Each point in the following plot is for a gene.)
The correlation between results for the same 7mer seed were higher (0.26), providing another example of the Iron Law of RNAi Screening. (Each point in the following plot is for a 7mer seed.)
It is also worth noting that the seed-based correlation would likely have been much higher, had the Dharmacon siRNAs been screened individually (see details below).
Conclusion
The only effective way to avoid off-target effects in RNAi screening is to use high-complexity reagents like siPOOLs, which dilute away off-target effects while maintaining strong on-target silencing.
Analysis details
To calculate the Ambion by-gene value, the mean PPT value was taken for the 3 on-target siRNAs for the gene. (The Dharmacon pooled library only has 1 value per gene, so no further calculation is necessary.)
To calculate the Ambion by-seed value, the mean PPT value was taken for all siRNAs with the 7mer. For Dharmacon, the pool value was assigned to each siRNA, and then siRNAs were grouped by their 7mer seed in order to calculate the seed mean. This means that the Dharmacon siRNA seed value is actually the average from 4 different siRNAs (with different seeds). Had the Dharmacon siRNAs been screened individually, the correlation with Ambion seed results would have been higher.
This blogpost describes issues encountered in target validation and how to safeguard against poor reproducibility in RNAi experiments.
The importance of target validation
More than half of all clinical trials fail from a lack of drug efficacy. One of the major reasons for this is inadequate target validation.
Target validation involves verifying whether a target (protein/nucleic acid) merits the development of a drug (small molecule/biologic) for therapeutic application.
Failing to adequately validate a target can burden a pharma with roughly 800 million to 1.4 billion in drug development costs. Impact is not only monetary as largesite closures often result as companies struggle to save costs and a reduced production effort deprives patients of new medicines.
Performing target validation well
Special attention should therefore be given to performing target validation techniques well.
Overview of target validation techniques (Lindsay, Nat Review Drug Discovery, 2013)
Many of these techniques involve inhibiting target expression to establish its relevance in a cellular or animal disease model. This can be performed with chemical probes, RNA interference (RNAi), genetic knock-outs, and even targeted protein degradation.
The reproducibility of these techniques however has been an issue of concern for drug developers. Less than half of all findings from peer-reviewed scientific publications was reported to be successfully reproduced.
Dismal rates of reproducibility from several pharma-led cancer-focused studies ranged from 11% (Amgen) to 25% (Bayer). A review by William Kaelin Jr sums up the common pitfalls of preclinical cancer target validation. One of his key points:
Cellular phenotypes caused by a chemical or genetic perturbant should be considered to be off-target until proved otherwise, especially when the phenotypes were detected in a down assay and therefore could reflect a nonspecific loss of cellular fitness. It is only by performing rescue experiments that one can formally address whether the effects of a perturbant are on-target.
The comment highlights the issue of reagent non-specificity as a notable contribution towards poor reproducibility.
Certainly, for RNAi the wide-spread off-target effects of siRNAs has been observed in numerous publications. The mechanism being well-established to be based on microRNA-like seed-based recognition of non-target genes. The effect dominates over on-target effects in many large RNAi screens, illustrating the depth of the problem.
Reagent non-specificity is not restricted to RNAi. There have been multiple reports of non-specificity for gene editing technique, CRISPR, which can be read about in detail here, here and here. Recent publications continue to shed more light on its potential off-targets as we learn more about this relatively new technique.
Even chemical probes may have multiple targets. It is hence imperative that more than one target validation technique be used to avoid confirmation bias.
Target validation – a story from Pharma
Back in 2013, when siTOOLs was just starting out, a pharma approached us with a target validation problem.
They were obtaining different results with 3 different siRNAs in a cellular proliferation assay. Despite all 3 siRNAs potently downregulating the target gene, they produced different effects on cell viability.
Which siRNA tool to trust?
Three different siRNAs against the same target were tested in a cell proliferation assay. Despite all 3 siRNAs showing potent target gene silencing, effect on cell proliferation differed greatly.
A whole-transcriptome expression analysis performed for the 3 siRNAs and a siPOOL designed against the same target revealed the reason for the large variability.
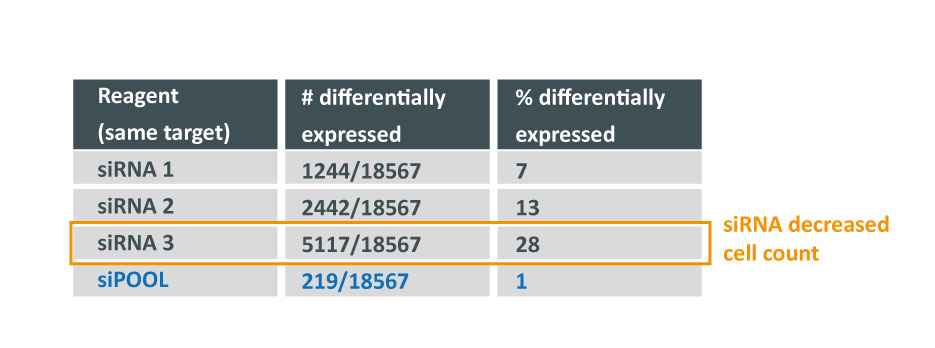
How many genes can you affect with an siRNA? Whole transcriptome analysis by microarray was performed and number and % of up and down-regulated genes are shown over total number of genes assayed (18567).
Despite all siRNA tools affecting the same target, the difference in extent of gene deregulation was astounding. With the greatest number of off-target effects, it was not surprising that siRNA 3 showed an impact on cell proliferation.
In contrast, siPOOLs had 5 to 25X less differentially expressed genes compared to the 3 commercial siRNAs against the same target. An expression analysis carried out for another gene target showed similar results i.e. siPOOLs having far less off-targets.
The target was dropped from development. A great example where failing early is a good thing, though it was not without costs from validating the multiple siRNAs.
The recommended target validation tool
Functioning like a pack of wolves, siPOOLs increase the chances of capturing large and difficult prey, while making full use of group diversity to compensate for individual weakness.
siPOOLs efficiently counter RNAi off-target effects by high complexity pooling of sequence-defined siRNAs. This enables individual siRNAs to be administered at much lower concentrations, below the threshold for stimulating significant off-target gene deregulation. Due to having multiple siRNAs against the same target gene, target gene knock-down is maintained and in fact becomes more efficient.
siPOOLs increase targeting efficiency, avoiding knock-down variability. Figure shows rtqPCR quantification of target RNA levels when two siPOOLs vs two siRNAs against 36 genes were tested.
We still recommend using multiple target validation techniques. As a first evaluation however, siPOOLs are quick, easy and most of all, reliable.
Summary:Low-complexity siRNA pooling (e.g. Dharmacon siGENOME SMARTpools) does not prevent siRNA off-targets. It may in fact exacerbate off-target effects. Only high-complexity pooling (siPOOLs) can reliably ensure on-target phenotypes.
Low-complexity pooling increases the number of siRNA off-targets
One of the claims often made in favour of low-complexity pooling (e.g Dharmacon siGENOME SMARTpools) is that this pooling reduces the number of seed-based off-target effects compared to single siRNAs.
If this were true, we would expect different low-complexity siRNA pools for the same gene to give similar phenotypes. But this is not the case.
Published expression data shows that low-complexity pooling actually increases the number of off-targets.
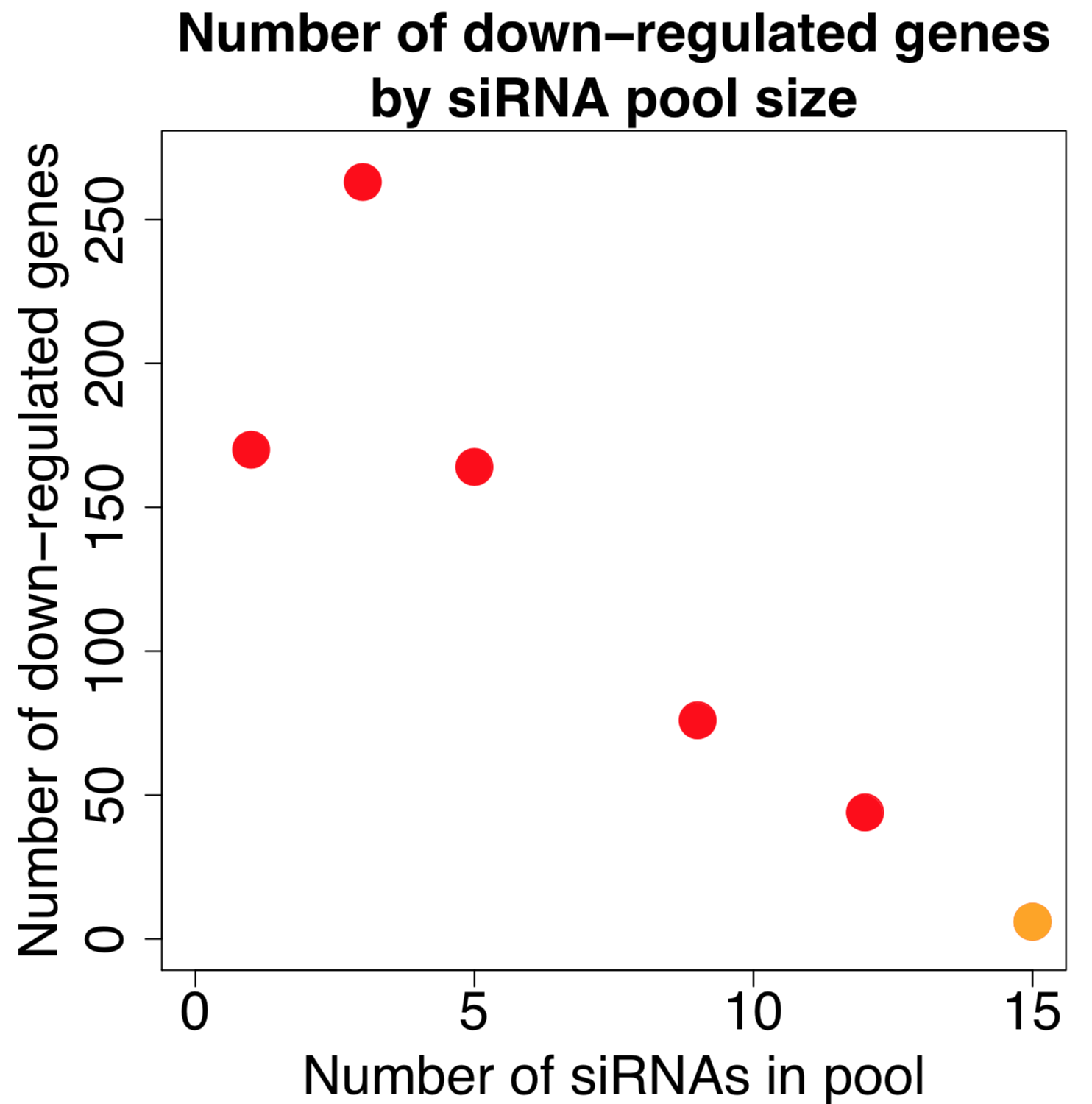
Kittler et al. (2007) looked at the effect of combining differing number of siRNAs in low to medium complexity siRNA pools (siRNA pools sizes were: 1, 3, 5, 9, and 12).
Their work showed that the number of down-regulated genes (50% or greater silencing) actually increases when small numbers of siRNAs are combined. Only when larger numbers of siRNAs are combined does the number of off-targets start to drop:
[The figure is based on data from GEO dataset GSE6807. Down-regulated genes are those whose expression is reduced by 50% or more. Note that the orange point is taken from our 2014 NAR paper, as we are not aware of other published expression datasets with this many pooled siRNAs. A few caveats with combining these datasets are that they use different target genes, siRNA concentrations, and the data comes from a different expression platform.]
Low-complexity pooling: a bad solution for siRNA off-targets
Low-complexity pooling does not get rid of the main problem associated with single siRNAs: seed-based off-target effects. Based the above analysis, it can make it even worse. It also prevents use of the most effective computational measures against seed effects.
Redundant siRNA Activity (RSA) is a common on-target hit analysis method for single-siRNA screens. It checks how over-represented the siRNAs for a gene are at the top of a ranked screening list. If a gene has 2 or more siRNAs near the top of the list, it will score better than a gene that only has a single siRNA near the top of the list. This is one way to reduce the influence of strong off-target siRNAs.
Correcting single siRNA values by seed medians has also been shown to be an effective way to increase the on-target signal in screens. This correction is not effective for low-complexity pools, since each pool can contain 3-4 different seeds.
Off-target based hit detection algorithms (e.g. Haystack and GESS) are also only effective for single-siRNA screens. The advantage of these algorithms is that it permits the detection of hit genes that were not screened with on-target siRNAs. These algorithms are not effective for low-complexity pool screens.
Our recommendation: do not convert single siRNAs into low-complexity pools, rather use high-complexity siPOOLs to confirm hits
We do not recommend that screeners combine their single siRNA libraries into low-complexity pools (e.g. combining 3 Silencer Select siRNAs for the same target gene). If possible, it is better to screen the siRNAs individually and then apply seed-based correction, RSA and seed-based hit-detection algorithms.
The time saved by only screening one well per target may prove illusory when the deconvolution experiments show that the individual siRNAs have divergent phenotypes.
It is probably better to deal with off-target effects up front (by screening single siRNAs) than to be surprised by them later in the screen (during pool deconvolution).
Summary: Conventional siRNAs have a high probability of giving off-target phenotypes. siRNA off-target effects can be reduced by using more specific reagents or narrowing the assay focus (to reduce the number of relevant genes). Even when the assay is relatively focused, more specific reagents significantly increase the probability of observing on-target effects.
Probability of siRNA off-target phenotype depends on reagent specificity and assay biology
The probability of getting an off-target effect from an siRNA depends on several factors, the main ones being reagent specificity and assay biology. If an siRNA down-regulates a large number of genes, or if an assay phenotype can be induced by a large number of genes, the probability of observing an off-target phenotype increases.
siRNAs can down-regulate many off-target genes
Garcia et al. (2011) compiled 164 different microarray experiments measuring gene expression following transfection with siRNAs. The mean number of down-regulated genes in these experiments was 132 and the median was 68 (down-regulated genes were silenced by 50% or more).
As noted in earlier studies of gene expression following siRNA treatment (e.g. Jackson et al. 2003), few of the down-regulated genes are shared between siRNAs with the same target gene. This suggests that the down-regulated genes are not the downstream result of target gene knockdown (i.e. they are mostly off-target).
High-complexity pooling of siRNAs (e.g. with siPOOLs) can reduce the number of down-regulated genes.
The following figure, based on data from Hannus et al. 2014, shows the difference between the gene expression changes caused by a single siRNA (left) and a high-complexity siRNA pool (siPOOL, right), which also includes that same single siRNA:
Estimating the probability of siRNA off-target phenotypes
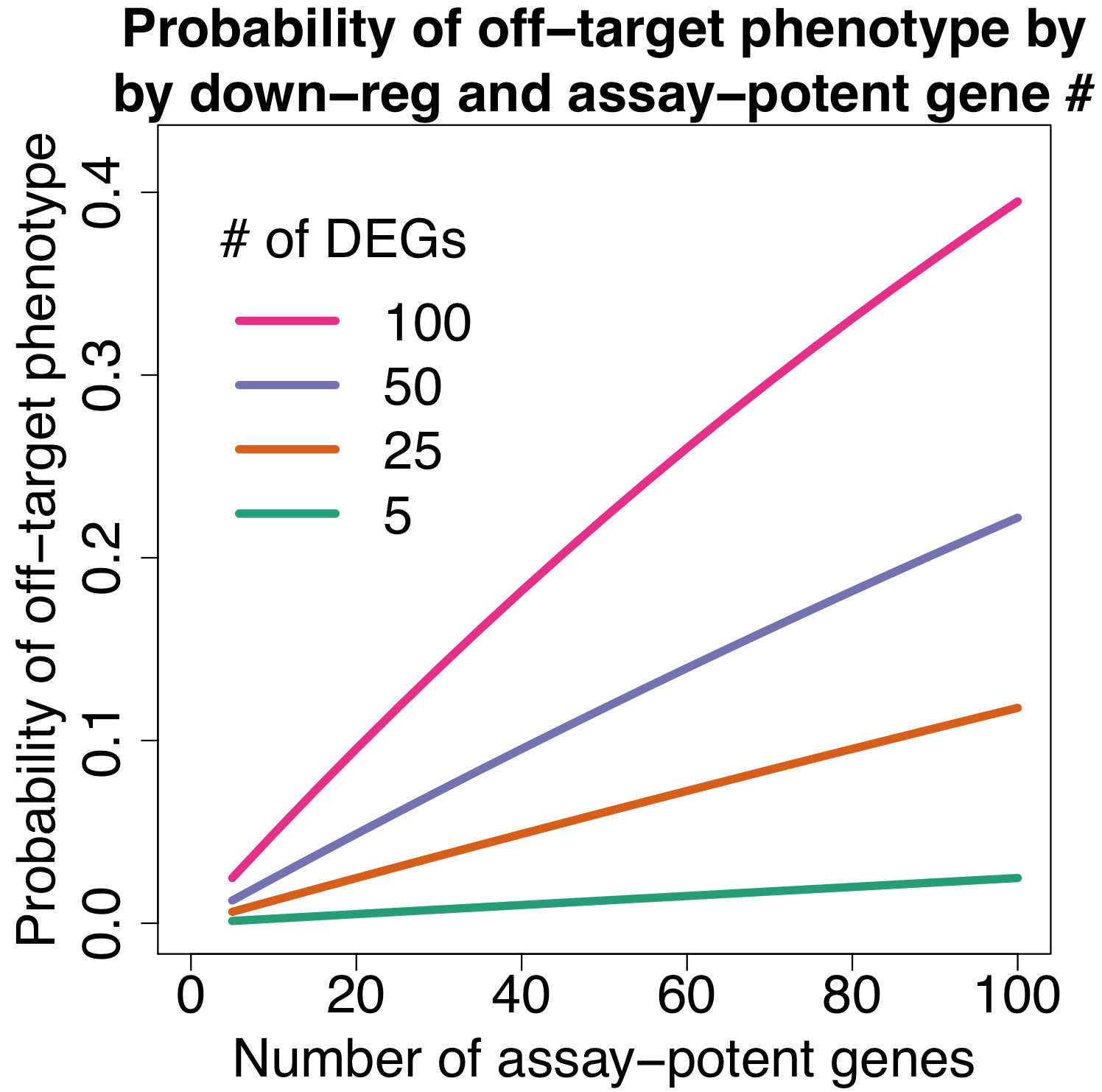
Assuming different numbers of down-regulated genes (off-target) and different numbers of potent genes involved in assay pathways, we can try to estimate the probability of an siRNA giving an off-target effect.
The following plot shows the probability of getting an off-target effect when:
down-regulated means that gene expression is reduced by 50% or more
in the Garcia paper dataset, the mean is 132 and median is 68
assuming different numbers of assay-potent genes
an assay-potent gene is one whose down-regulation by 50% or more is sufficient to produce a hit phenotype
for assays with more general phenotypes (e.g. cell count) we would expect more assay-potent genes
We can see that even if there are only 20 assay-potent genes, there’s a nearly 10% chance of getting an off-target phenotype when siRNAs down-regulate 100 off-target genes (which is close to the average observed in the Garcia dataset).
In a genome-wide screen of 20,000 genes with 3 siRNAs per gene, we would thus expect 2,000 off-target siRNAs.
In contrast, a more specific reagent that only down-regulates 5 off-target genes only has a 0.5% change of producing an off-target phenotype. For the above-mentioned genome-wide RNAi screen, we would expect only 100 off-target siRNAs (a 20-fold reduction).
The importance of RNAi reagent specificity
The above analysis demonstrates the importance of using specific siRNA reagents.
Changing an assay to make the phenotypic readout narrower (to reduce the number of genes capable of inducing a phenotype) is one way to reduce the risk of off-target phenotypes. But this may be a lot of work and is not necessarily desirable or even possible.
A more ideal solution is the use of a specific RNAi reagent, like siPOOLs.
postscript
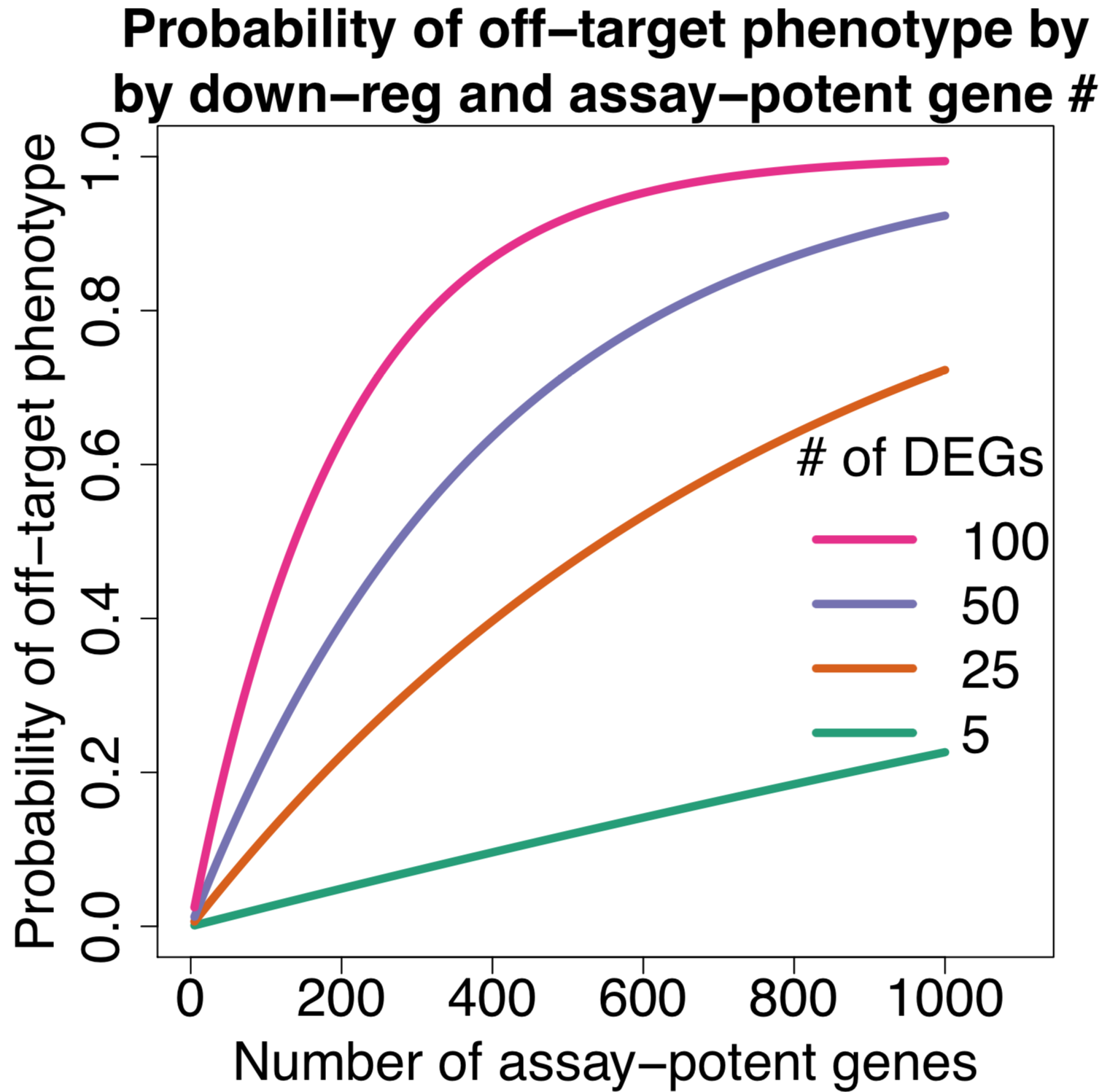
As the number of assay-potent genes increases, the probability of getting an off-target phenotype approaches one.
The following plot (same format as the one above) shows the distribution
The p-values were calculated using the hypergeometric distribution, assuming a population size of 20,000 (the approximate number of protein-coding genes in the human genome).
Note that one of the major simplifying assumptions of the above analysis is that all siRNAs have the same number of down-regulated off-target genes.
5 factors to consider in multi-gene targeting RNAi screens
Summary: Effective functional genomic screening depends on a variety of factors that need to be simultaneously addressed to obtain meaningful results. A recent Cell Reports paper demonstrates this by taking a holistic approach to siRNA screening with the use of multi-isoform/multi-gene targeting to address redundant paralogs and pathways in cancer cells.
The case for multi-gene targeting
Many RNAi screens use arrayed single gene knockdowns to find genes that play an important role in a biological process. The idea is that a single bullet is enough to take down its target leaving a gaping hole that one cannot fail to notice. In some cases, this is true, and is certainly relied upon by drug developers seeking to create specific mono-target drugs. However, in complex diseases like cancer, cells have evolved fail-safe mechanisms to make them more resistant to external assaults. A single bullet is simply not enough.
Take for example oncogenic protein RAF or Rapidly Accelerated Fibrosarcoma, a tyrosine kinase effector that is a component of the MAPK signalling pathway (Ras-Raf-MEK-ERK). RAF has three isoforms – ARAF, BRAF and RAF1 (also called CRAF). Studies in mouse embryonic development show they all share some form of functional redundancy as knocking out two isoforms produces more severe effects than knocking out each isoform alone.
Screens that target single genes/isoforms therefore tends to bias results towards genes that have no paralogs or only have single isoforms. This was indeed the reason why classical Ras effectors were not identified in previous screens.
Factors to consider in a multi-gene targeting RNAi screen
Determining gene combinations that make sense
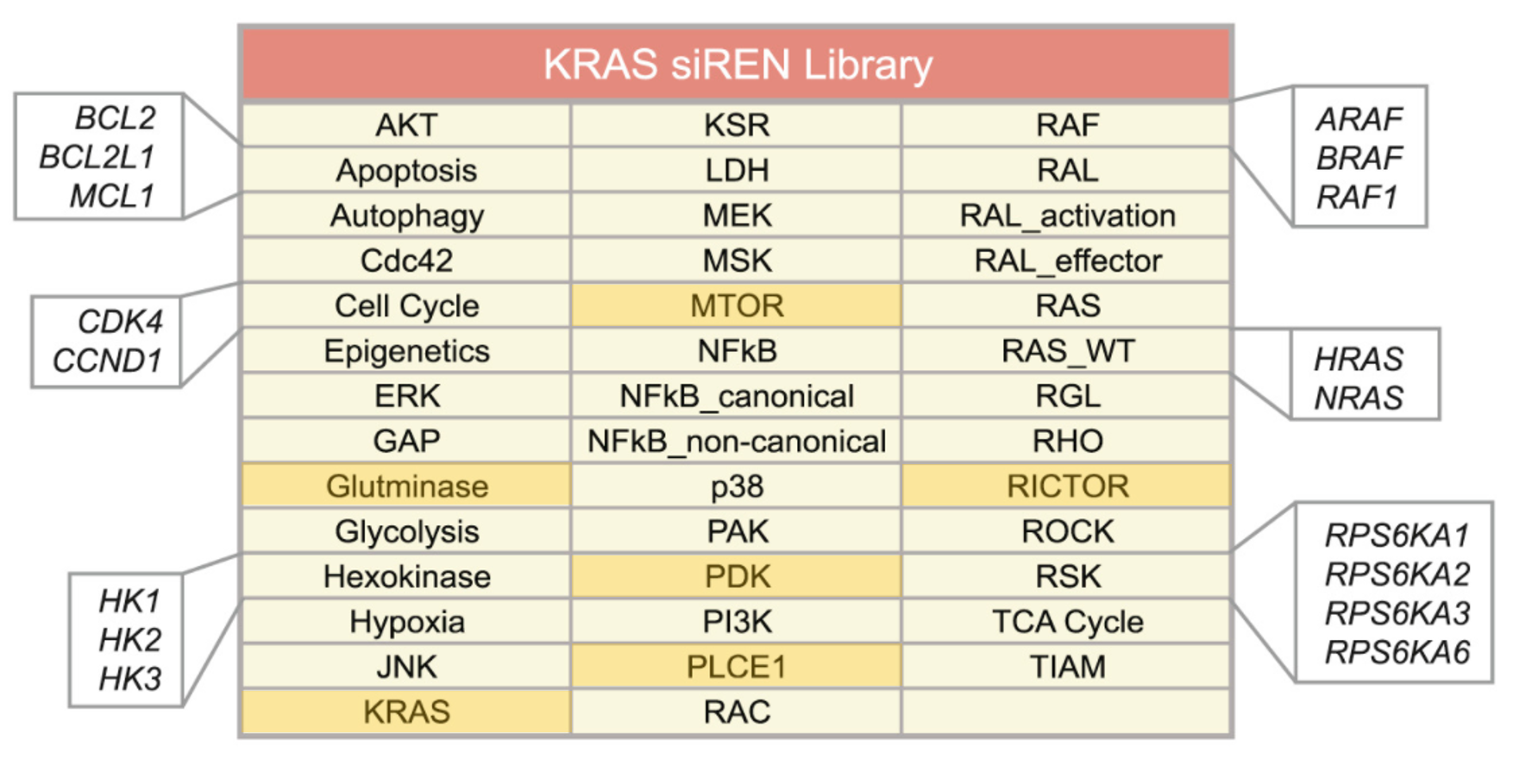
The authors of the study did a focussed siRNA screen on 41 RAS effector nodes represented by 84 genes. Out of the 41 nodes, 25 of them had 2-4 functional paralogs where combinatorial gene silencing was carried out with multiple siRNAs. 5 nodes knocked down multiple members of a protein complex. 5 nodes had siRNAs targeting multiple steps within a pathway. Only 6 nodes silenced single genes (highlighted).
The only caveat with designing such a screen is the requirement for prior knowledge to perform meaningful gene silencing combinations. In this instance, many of the Ras effector pathways are characterized sufficiently to do this well however in other less studied fields, this could be a challenge. Useful tools that would help in designing gene knockdown combinations would include pathway or phenotype databases such as KEGG, REACTOME or Wikipathways. The Phenovault which siTOOLs Biotech is developing, is yet another potentially useful tool.. more details to come!
Number and types of phenotypes
The authors also highlight how a screen that reads only one phenotype might miss other important gene functions. Many RNAi screens sadly still stick to measuring cell proliferation as their only read-out which is greatly influenced by siRNA off-target effects. Here, 5 different phenotypes were measured (cell size, proliferation, apoptosis, reactive oxygen species [ROS], and viability). It was noted that silencing of Cdc42 had little effect on cell viability yet a prominent effect on ROS levels.
To take this up a notch, analysis was also performed at the single-cell level in cells expressing uniform levels of GFP and co-transfected with GFP siRNA. This allowed authors to correlate phenotypes with levels of gene knockdown, generating dose-response curves. How clever!
A lot more work, but adds to data robustness especially when using single siRNAs that are known to be rather variable.
Heterogeneity of cell lines
Many reports and our own observations attest to the heterogenous response of different cell lines to the same treatment. In cancer especially, the large heterogeneity necessitates the use of multiple cell lines. Not doing so would be failing to account for the large genetic diversity observed in the clinic. The authors screened 92 cell lines derived from lung, pancreas and colorectal tissue.
Despite seeing heterogenous responses to node knockdowns, phenotypic responses could be distinguished into several groups based on effector engagement. A major group dependended on RAF through direct binding with KRAS, a second major group worked via RSK p90 S6 kinases to drive RSK-mTOR signalling. And a third minor group was dependent on RalGDS. They went on to focus on the first two major groups, naming them KRAS-type and RSK-type respectively.
Reagents – choosing siRNAs and siRNA concentrations
The authors used previously characterized siRNAs to select for more potent siRNAs. This involved an RNAi sensor reporter-based assay that required the generation of 20,000 clones. The reporter was also shRNA-based. Due to heterogeneity in Dicer-mediated cleavage of shRNA, its uncertain if knockdown potency is accurately reflected when translated to siRNAs (read about the difference between shRNAs and siRNAs).
In any case, its a lot of work to characterize all siRNAs to be used in a screen. Furthermore, off-target effects are not addressed.
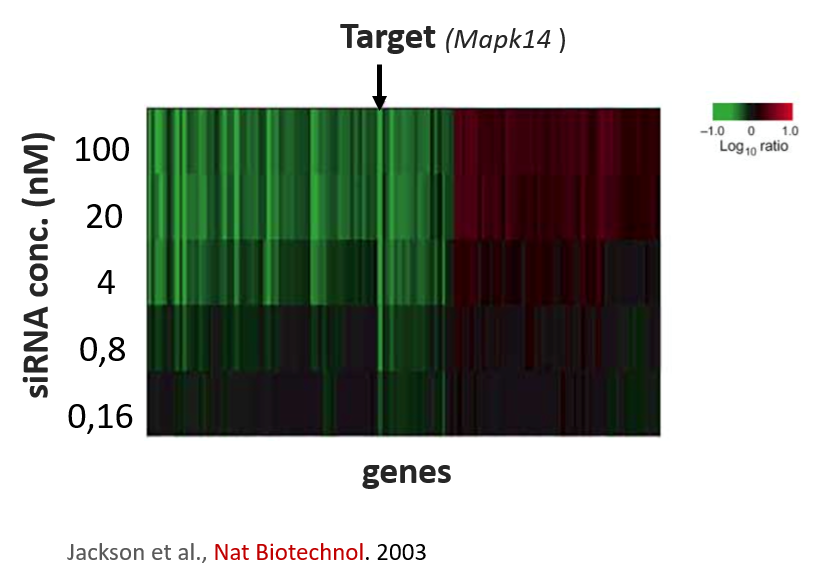
The authors stuck to a maximal concentration of 12 nM where 2 nM of siRNA was applied per gene. At 2 nM per siRNA, one still risks deregulating other genes. One of the first papers by Aimee Jackson et al., demonstrated an siRNA targeting MAPK14 deregulated many other genes even at concentrations of 1-4 nM.
An important consideration is to ensure total siRNA concentrations are kept constant. In which case, a negative control siRNA has to match or follow the maximal siRNA concentration used. Using different levels of siRNAs runs the risk of biasing off-target effects towards sequences present at higher concentrations.
To learn what the causes, extent and consequences of siRNA off-target effects are, read siTOOLs Technote 1)
Validating results
As with all scientific hypothesis, it helps to arrive at the same conclusion with different approaches.
The two different effector response subgroups identified also responded differently to small molecules. The KRAS-type lines being more sensitive to EGFR and ERK inhibition while the RSK-type lines more sensitive to inhibitors of PDK1, RSK, MTOR, S6K1 and DNA repair enzymes. This was attributed to the latter’s higher basal metabolic activity manifested in larger investments towards oxidative phosphorylation and mitochondrial ribosome maintenance.
By also projecting signatures obtained from cell lines into patient samples (in The Cancer Genome Atlas, TCGA), the subtypes were also effective at predicting differential sensitivity to multiple drug treatments. This highlights the importance in designing effective drug combinations in cancer.
Interestingly, the authors also performed CRISPR pooled screens in parallel. However, due to the restraints of being only able to knockout 1 gene at a time, smaller effects were seen due to gene redundancy. However, they did go on to use CRISPR as well to mutate key genes to affirm the pathway relationships established.
siPOOLs have been used successfully for multi-gene targeting for up to 4 genes, and potentially more. They also safely address off-target effects by high complexity pooling, enabling each siRNA to be applied at picomolar concentrations. For more articles on multi-gene targeting, read an older blogpost:
Summary: To address the question of whether one should avoid microRNA binding sites during siRNA design, we examined whether removing siRNAs that share seeds with native microRNAs would reduce the dominance of seed-based off-target effects in RNAi screening.
siRNA design and native microRNA target sites
Recently, we discussed a review of genomics screening strategies. The authors state:
RNAi screens are powerful and readily implemented discovery tools but suffer from shortcomings arising from their high levels of false negatives and false positives (OTEs) as can be seen when comparing the low concordance among the candidate genes detected in different screens using the same species of virus, e.g., HIV-1, HRV, or IAV (Booker et al., 2011; Bushman et al., 2009; Hao et al., 2013; Perreira et al., 2015; Zhu et al., 2014).
To address these concerns, improvements in the design and synthesis of next-gen RNAi library reagents have been implemented including the elimination of siRNAs with seed sequences that are complementary to microRNA binding sites.
Given that off-target effects via microRNA-like binding are the main source of RNAi screening phenotypes, avoiding native microRNA sites during siRNA design seems like a reasonable strategy. But does it make much difference in actual RNAi screens?
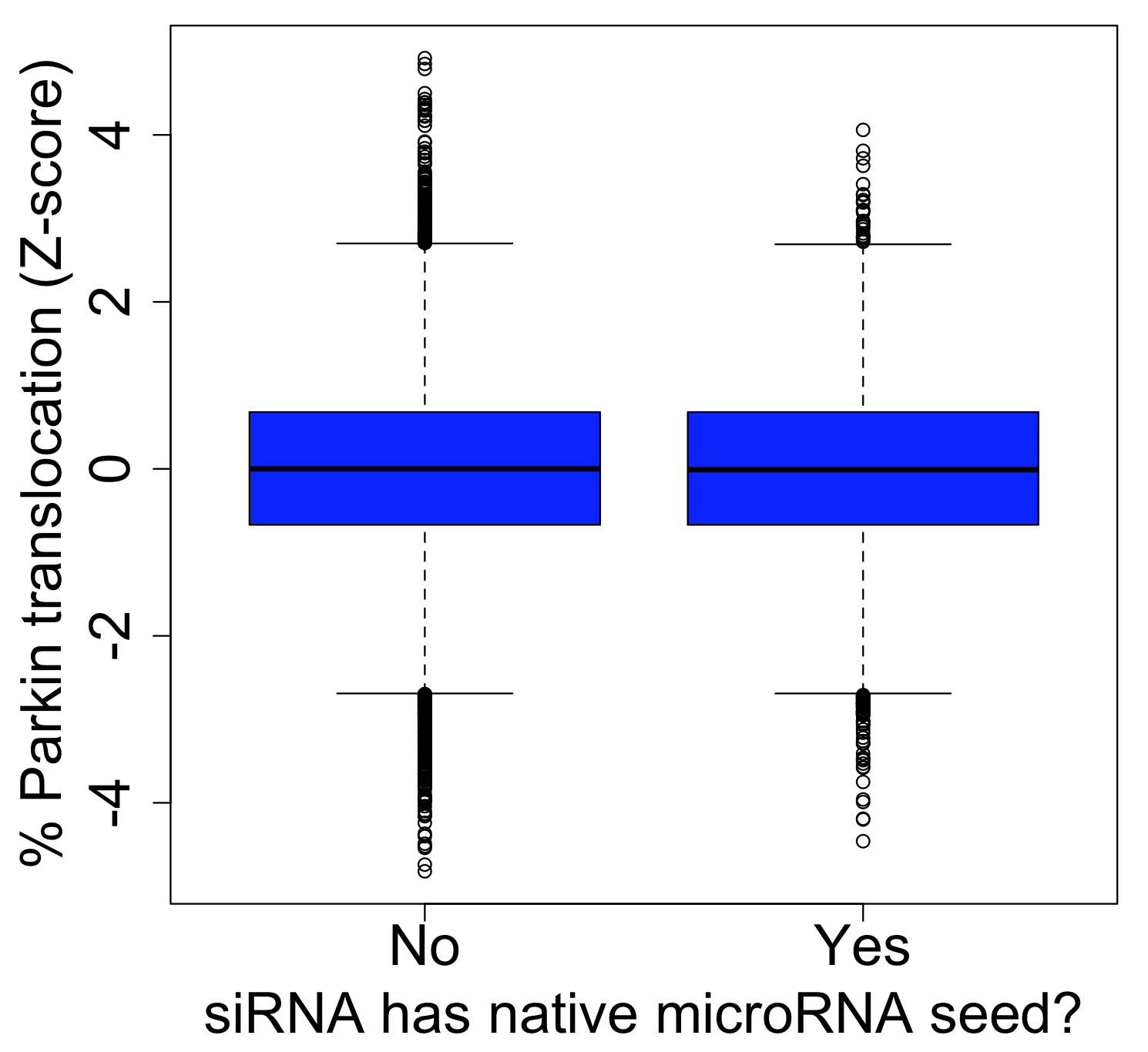
Hasson et al. 2013 performed a mitophagy screen using the Silencer Select siRNA library. About 12% of the ~65,000 screened siRNAs have a 7-mer seed shared by a miRBase microRNA.
The screen’s main phenotypic readout, % Parkin translocation (PPT), is strongly affected by seed effects. The intra-class correlation for siRNAs with the same seed is ~.51 (versus ~.06 for siRNAs with the same target gene). There appears to be no difference between how siRNAs with or without microRNA seeds behave:
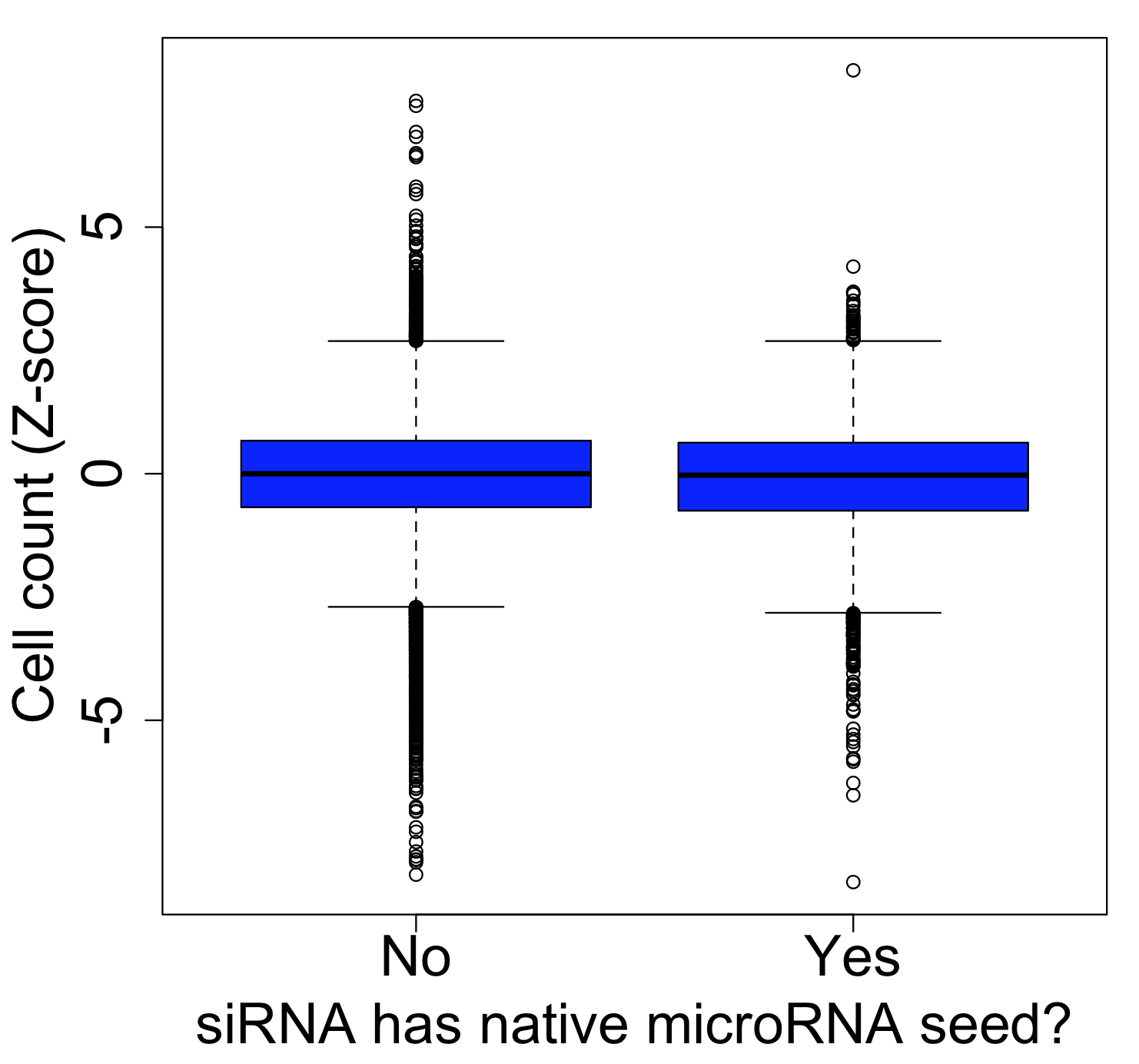
The same thing is found if we look at a less specific phenotype like cell count (which should be more broadly susceptible to off-target effects, as more genes should affect this phenotype):
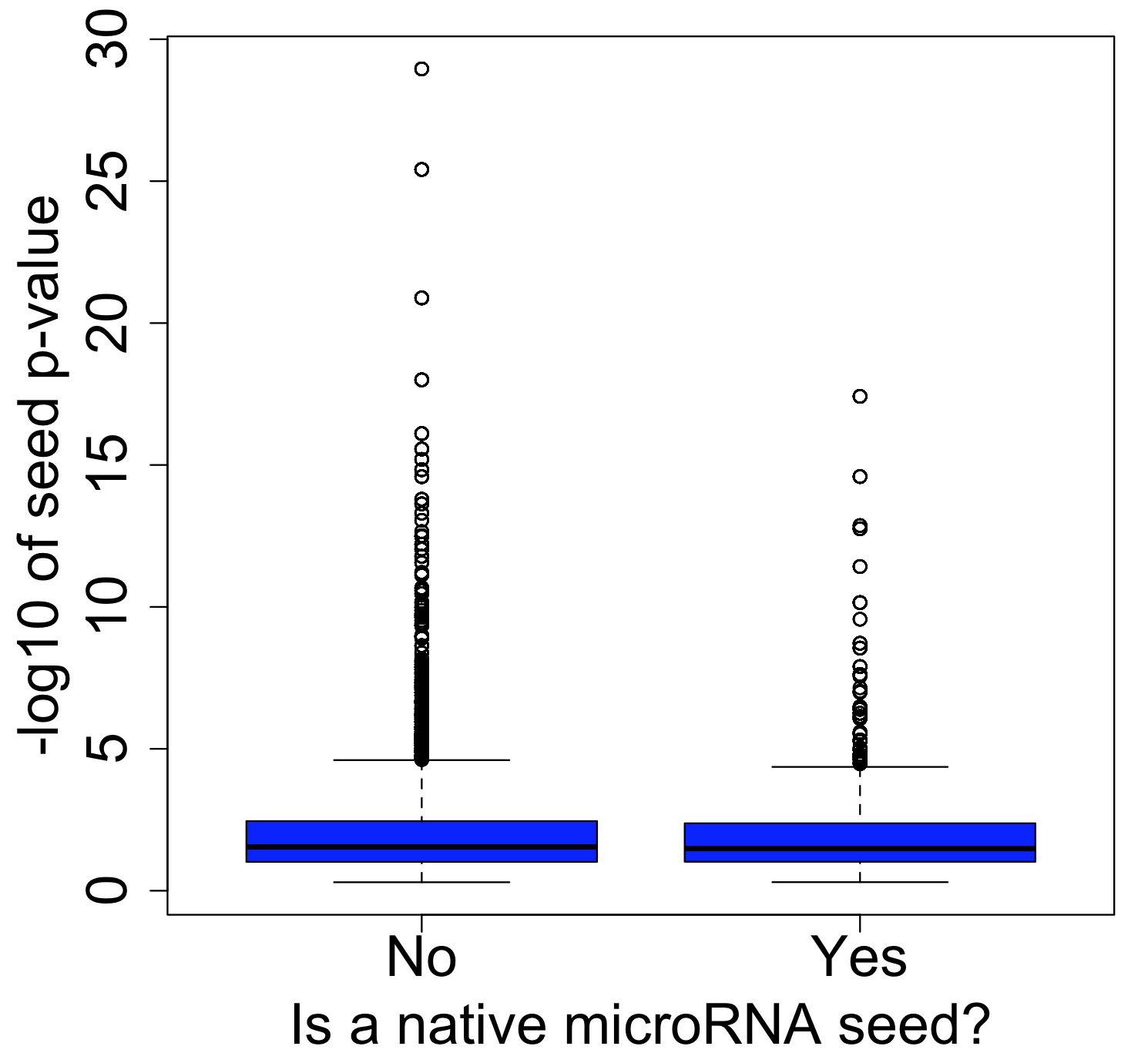
And if we look at seeds that are enriched at the top of the screening list (sorted by descending PPT), we also don’t see much difference between siRNAs with or without native microRNA seeds. (Note that the seed p-value is calculated in a similar way to RSA, based on how over-represented a seed is towards the top of a ranked list)
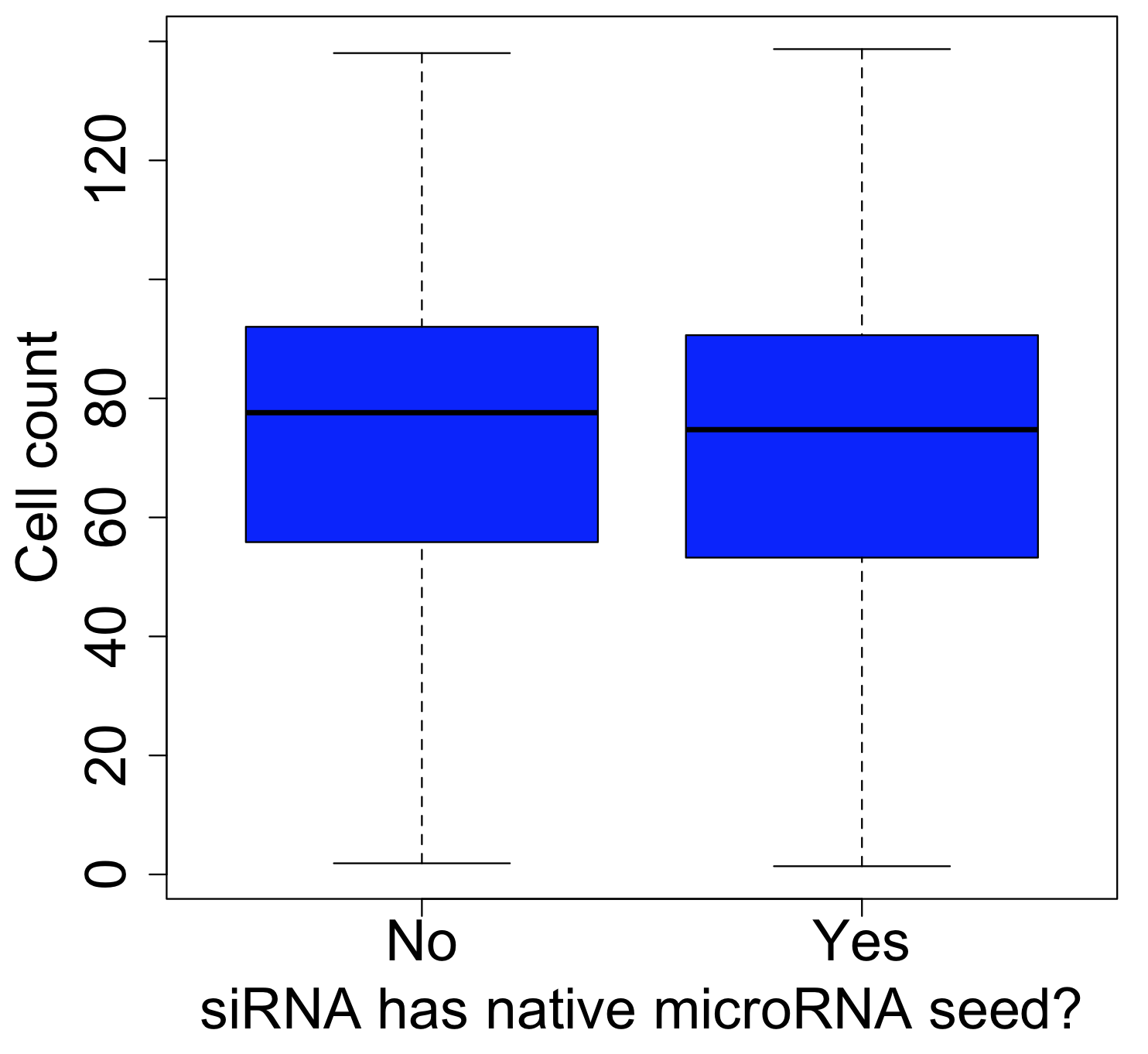
We also examined a general phenotypic readout (cell viability) in a dozen large-scale RNAi screens.
For some screens, we do see a slight shift in the values for siRNAs with or without native microRNA seeds.
For example, a genome-wide screen of Panda et al. 2017 (also using the Silencer Select library) shows a slight decrease in viability for siRNAs with native microRNA seeds:
Removing those siRNAs does not change the dominance of seed-base off-targets.
The intra-class correlation (ICC) for siRNAs with the same 7-mer seed is ~.53, with or without the inclusion of siRNAs with native microRNA seeds, while ICC for siRNAs with the same target gene is only ~.06.
Coming back to the quote from the review article on genomic screening, next-gen RNAi library reagents that avoid native microRNA seeds are not expected to be much better than siRNAs that include them.
The most effective way to avoid seed-based off-target effects is to use high-complexity siRNA pools (siPOOLs). Learn more about siPOOLs
Disrupting lncRNA function with siPOOLs (RNAi), antisense oligos and CRISPR
This blogpost covers methods used in the disruption of lncRNA function. Specifically focusing on RNA interference (with siPOOLs), antisense oligos, and CRISPR approaches. Challenges faced with these approaches are addressed.
Long non-coding RNAs (lncRNAs) make up a major subgroup of RNAs and are defined as over 200 nucleotides long with limited protein-coding potential. There are three times as many genes producing lncRNAs as opposed to proteins. Numerous studies have described functional roles of lncRNAs in development and disease. This has stimulated major global interest and intense efforts to decode lncRNA function.
Disrupting lncRNA function
One way to find out what a lncRNA does is to decrease its expression, thereby disrupting its function. Current methods of downregulating lncRNA expression include knockdown approaches with siRNA and antisense oligos (ASOs), or knockout approaches with CRISPR, TALENs and other techniques involving DNA nucleases.
As we have mentioned before, knockdown and knockout approaches employ different mechanisms and as a result sometimes yield different results. Hence it is highly recommended to employ both techniques when possible to thoroughly validate lncRNA function.
LncRNA functional knockdown – RNAi and antisense approaches
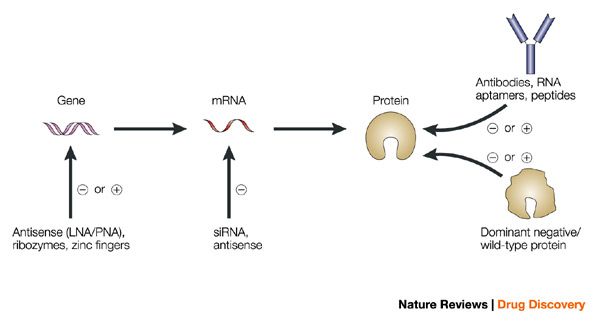
LncRNA knockdown involves the transient downregulation of lncRNAs at the RNA level. This typically involves RNA degradation mediated by the RNA interference (RNAi) machinery for siRNAs, or with RNase H for ASOs.
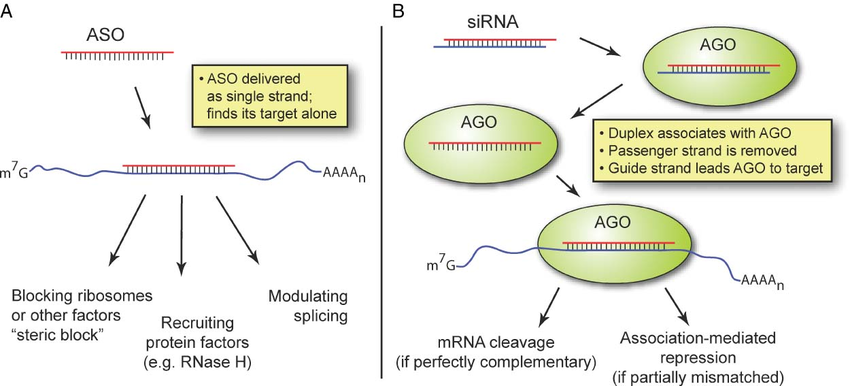
How ASOs and siRNAs downregulate RNA
Figure from Watts, J. K. & Corey, D. R. Silencing disease genes in the laboratory and the clinic. J. Pathol. 226, 365–79 (2012).
Some challenges that both technologies face when targeting lncRNAs:
low endogenous expression of lncRNA may limit efficiency of knockdown
accessibility of siRNA/ASO to lncRNA may be limited by secondary structure (created by folding of the lncRNA and self-base pairing)
accessibility to siRNA/ASO to lncRNA may be limited by bound proteins
off-target effects
Does cellular localization matter when disrupting lncRNA function?
Cellular localization of lncRNAs was reported to account for differences in knockdown efficiency by ASOs compared with siRNAs. Although there have been observations that RNAi factors are present in the nuclei, siRNAs were reportedley less efficient than ASOs for modulating nuclear-localized lncRNAs (Lennox and Behlke, Nucleic Acids Res, 2016).
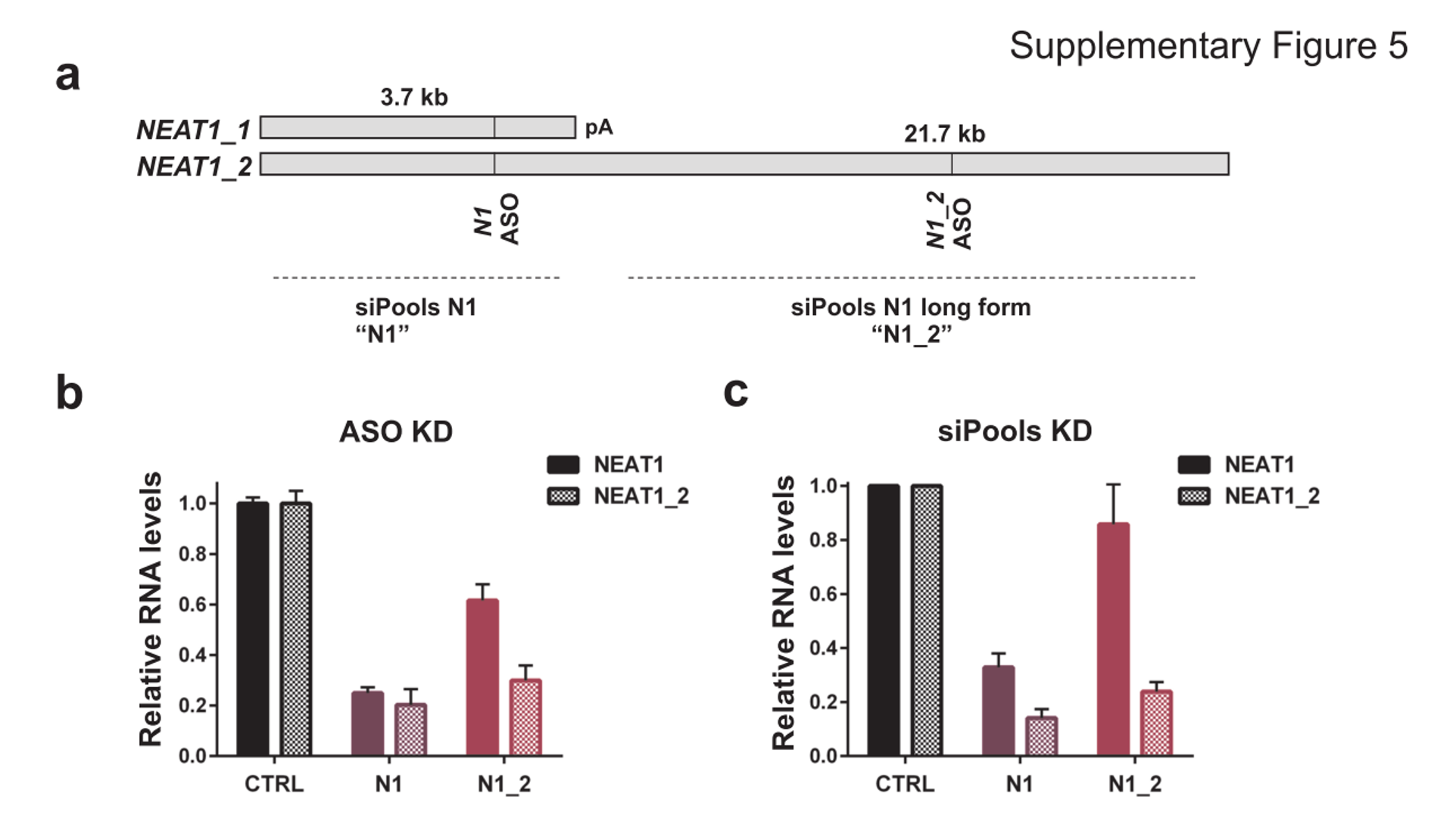
This does not appear to apply to all cases as using siPOOLs (high complexity pooled siRNA) or ASOs led to similar downregulation of NEAT1, a nucleus-localized lncRNA:
Downregulation of lncRNA NEAT1 with siPOOLs and ASOs
NEAT1 lncRNA has two isoforms, 3.7kb NEAT1_1 and longer 21.7kb NEAT1_2. MCF7 cells were transfected with either LNA GapmeRs (ASOs) or siPOOLs that target both isoforms (N1) or the long form only (N1_2). RNA levels of both isoforms (NEAT1) or only the long isoform (NEAT1_2) were quantified after 24h. (Adriaens et al., Nat Med, 2016)
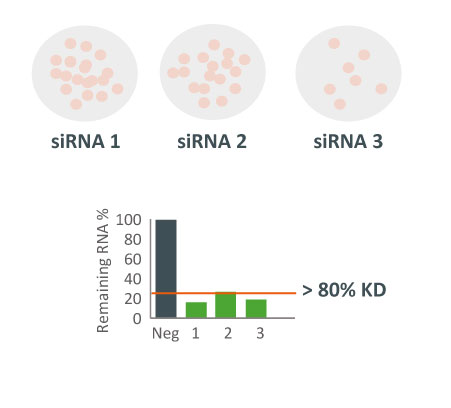
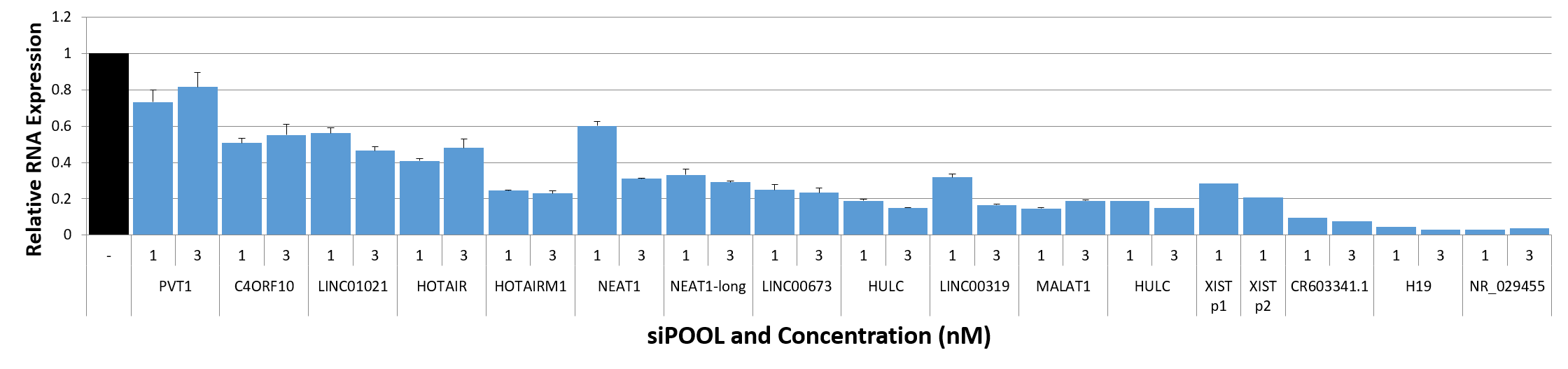
siPOOLs also worked well for XIST and MALAT1 (~80% KD at 1 nM), both nuclear-localized lncRNA. Notably however, cytosolic-localized lncRNAs such as H19 were much more efficiently targeted with the high complexity siRNA pools (> 95% KD at 1 nM).
siPOOL knockdown efficiency of lncRNAs
siTOOLs Biotech in-house data showing knockdown efficiencies of siPOOLs against 16 lncRNAs tested at 1 or 3 nM in standard cell lines (MCF7, A549, Huh7). Assayed by real-time quantitative PCR after 24h.
Compared to coding genes, the above-mentioned factors do limit efficiencies of knockdown approaches. But with siPOOLs, the greater diversity of siRNA sequences is expected to increase chances of association with the target RNA. In-house data shows 12 of 16 tested lncRNAs showed good knockdown efficiencies of > 70% with siPOOLs.
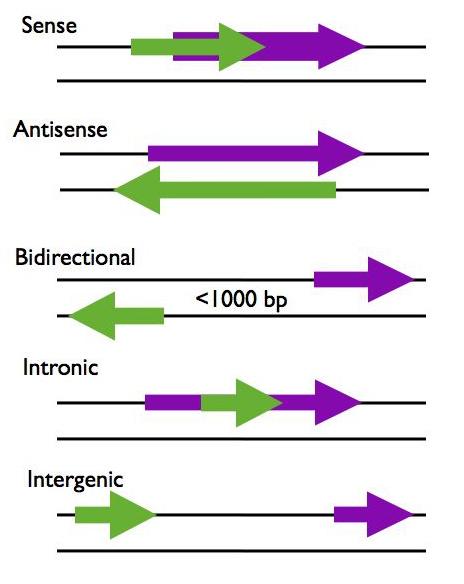
The genomic distribution of lncRNA loci is rather complex. They are typically categorized in relation to their proximity with protein coding genes.
Types of lncrna
Figure showing lncRNA loci in green and protein-coding loci in purple. Arrows indicate direction of transcription. Figure and description below from McManus lncRNA presentation: https://mcmanuslab.ucsf.edu/node/251
Sense – The lncRNA sequence overlaps with the sense strand of a protein coding gene.
Antisense – The lncRNA sequence overlaps with the antisense strand of a protein coding gene.
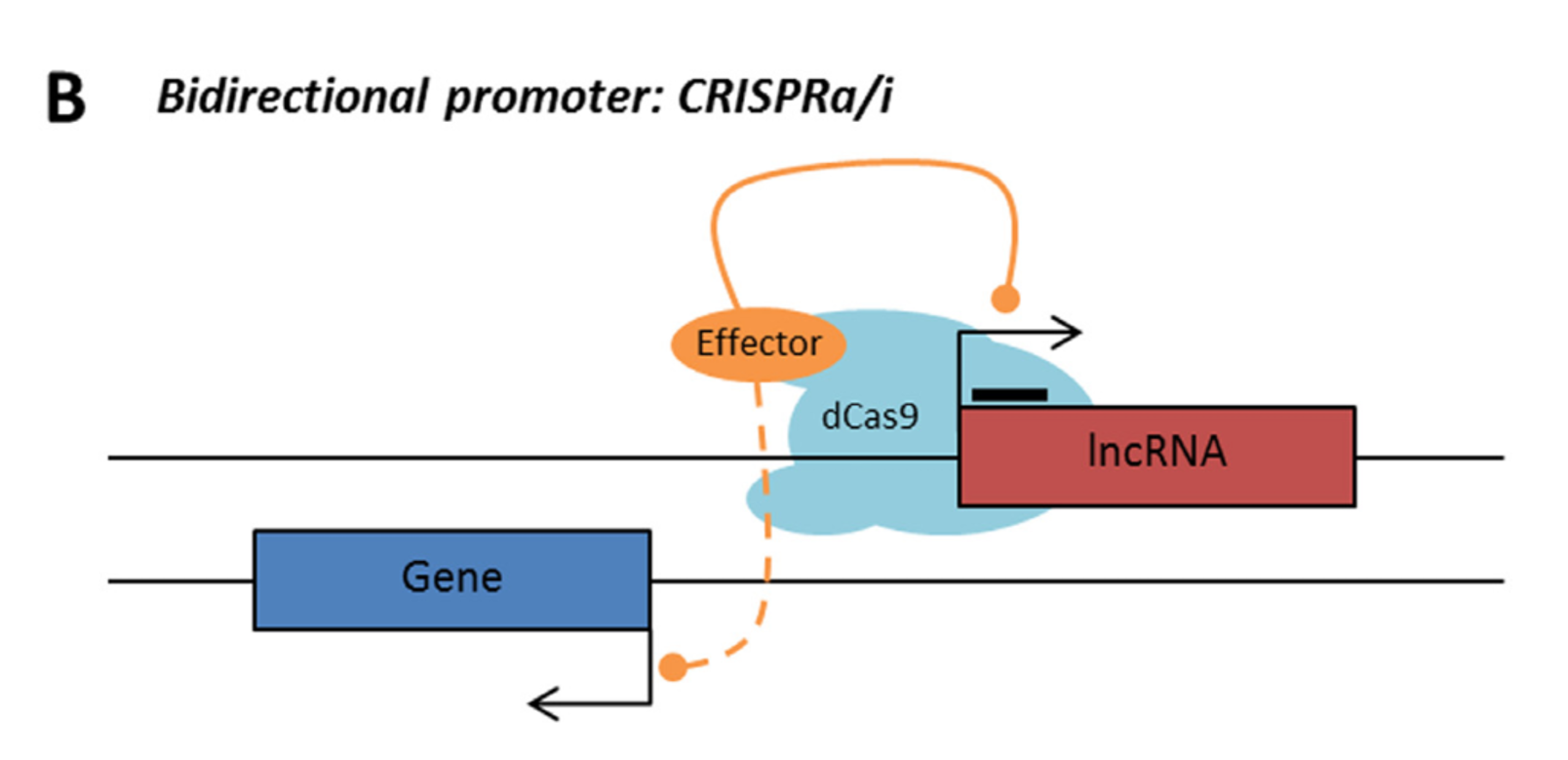
Bidirectional – The lncRNA sequence is located on the opposite strand from a protein coding gene whose transcription is initiated less than 1000 base pairs away.
Intronic – The lncRNA sequence is derived entirely from within an intron of another transcript. This may be either a true independent transcript or a product of pre-mRNA processing
Intergenic – The lncRNA sequence is not located near any other protein coding loci.
Hence disrupting lncRNAs with DNA nucleases can be a challenging affair that runs the risk of affecting neighbouring genes.
How many lncRNAs can be CRISPRed?
Goyal et al. 2017 performed a genome-wide “CRISPRability” analysis to evaluate the risks and utility of CRISPR for disrupting lncRNA function.
Introducing mutations with CRISPR is generally not applicable for lncRNAs. Mainly due to difficulty predicting active functional domains and the fact that some lncRNAs exert phenotypes through the act of transcription per se.
Deleting the entire lncRNA is an option but not when it overlaps with other genes. Hence, the major approach is to target lncRNA promoters. But then we run into the problem of affecting neighbouring genes that share promoters.
So they came up with three “CRISPRability” rules to avoid potential effects on neighbouring genes:
Rule 1: Sense, antisense and intergenic lncRNAs are considered “non-CRISPRable” when transcribed from bidirectional promoters, defined by presence of another promoter present 2000bp upstream/downstream of lncRNA start.
LncRNA with bidirectional promoter
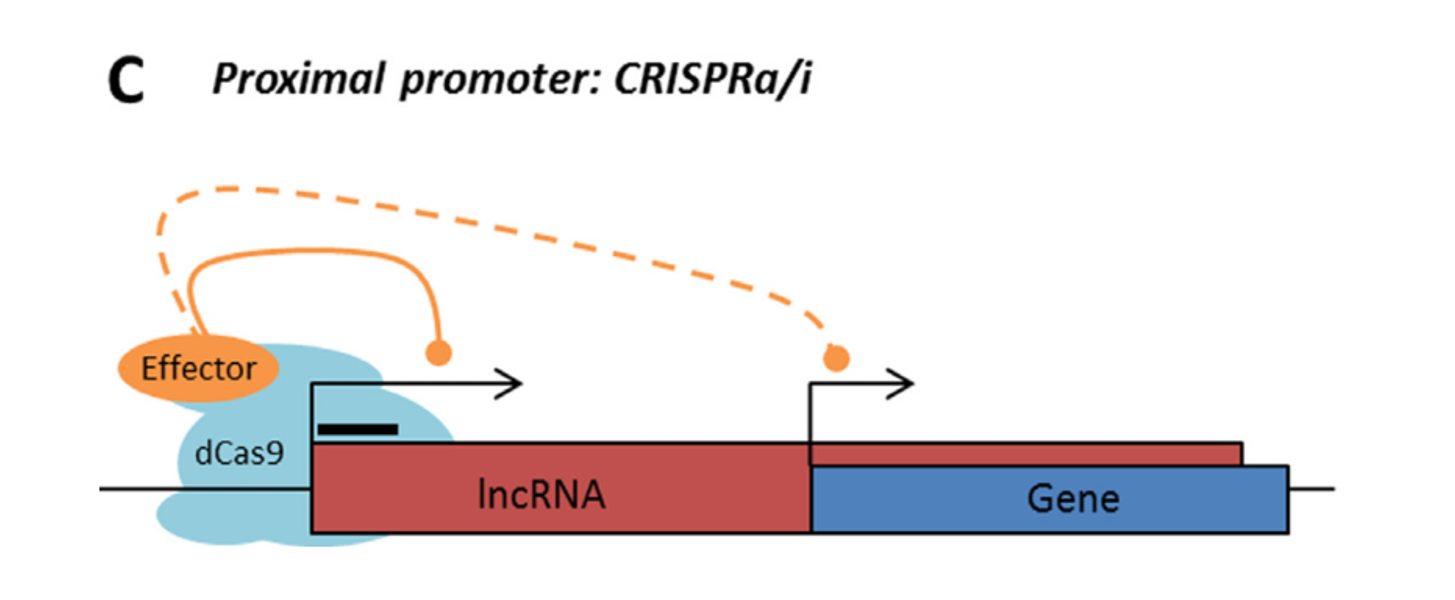
Rule 2: Sense, antisense and intergenic lncRNAs are considered “non-CRISPRable” when the start of the lncRNA is located closer than 2000p to the start of the neighbouring gene, excluding lncRNAs transcribed from bidirectional promoters – termed “proximal promoters“.
LncRNA with proximal promoter
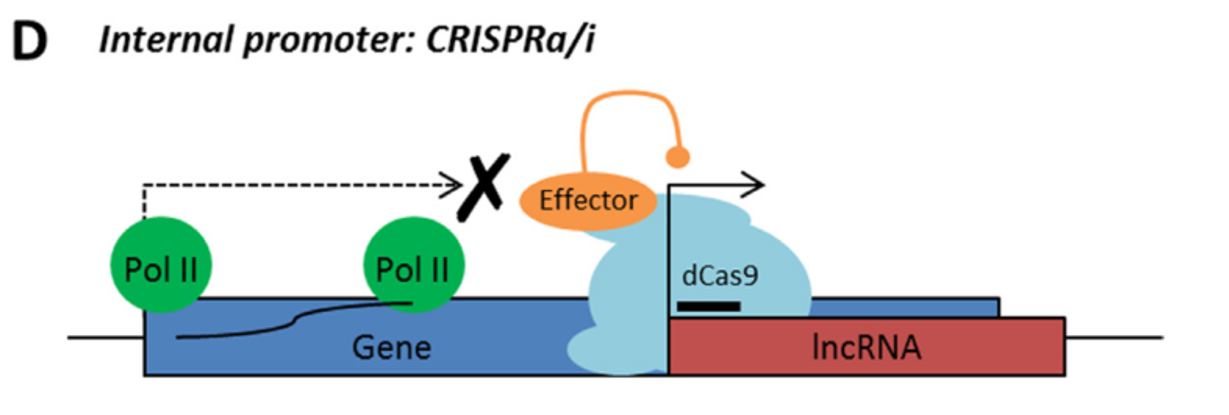
Rule 3: Sense and antisense lncRNAs are considered “non-CRISPRable” when transcribed from internal promoters, where the start of the lncRNA falls within the gene body of another coding/non-coding transcript. This would include intronic lncRNAs.
LncRNA with internal promoter
After applying “CRISPRability” rules, only 38% of all lncRNAs were suitable for CRISPR-based functional disruption
CRISPRability of lncRNAs
Figure from Goyal et al., 2017 showing proportion of lncRNAs that fall within the 3 rules of “CRISPRability”
The study went on to corroborate the relevance of the classification by testing effects of CRISPR/Cas9 compared to ASOs/siRNA on their targets and neighbouring genes.
HOTAIR downregulation by CRISPR and siPOOL
An example involved lncRNA HOTAIR that arises from the HOXC locus which regulates expression of several genes including HOXC11. They found that dCas9-KRAB , which produces CRISPR-based transient inhibition (CRISPRi) by blocking transcription, caused knockdown of HOXC11 when designed to target HOTAIR. This occurred for all 3 independent sgRNAs. siPOOL-mediated knockdown of HOTAIR, in contrast, did not affect HOXC11.
Similar scenarios were seen with coding genes, in particular for well-known tumour suppressor TP53, where neighbouring gene WRAP53alpha tended to be downregulated by dCas9-KRAB. This effect was absent with siPOOLs targeting TP53.
It therefore pays to carefully note the genomic neighbourhood of lncRNAs when using CRISPR for disruption. A careful scientist would also monitor the expression of neighbouring/overlapping genes in parallel to the target gene. Orthogonal methods such as RNAi (with siPOOLs), or rescue experiments that restore expression of the lncRNA, is recommended to fully evaluate lncRNA function.
Summary: Correcting for seed-based off-targets can improve the results from RNAi screening. However, the correlation between siRNAs for the same gene is still poor and the strongest screening hits remain difficult to interpret.
Seed-based off-target correction has little effect on reagent reproducibility
Given that seed-based off-targets are the main cause of phenotypes in RNAi screening, trying to correct for those effects makes good sense.
The dominance of seed-based off-targets means that independent siRNAs for the same gene usually show poor correlation.
If one could correct for the seed effect, the correlation between siRNAs targeting the same gene may improve.
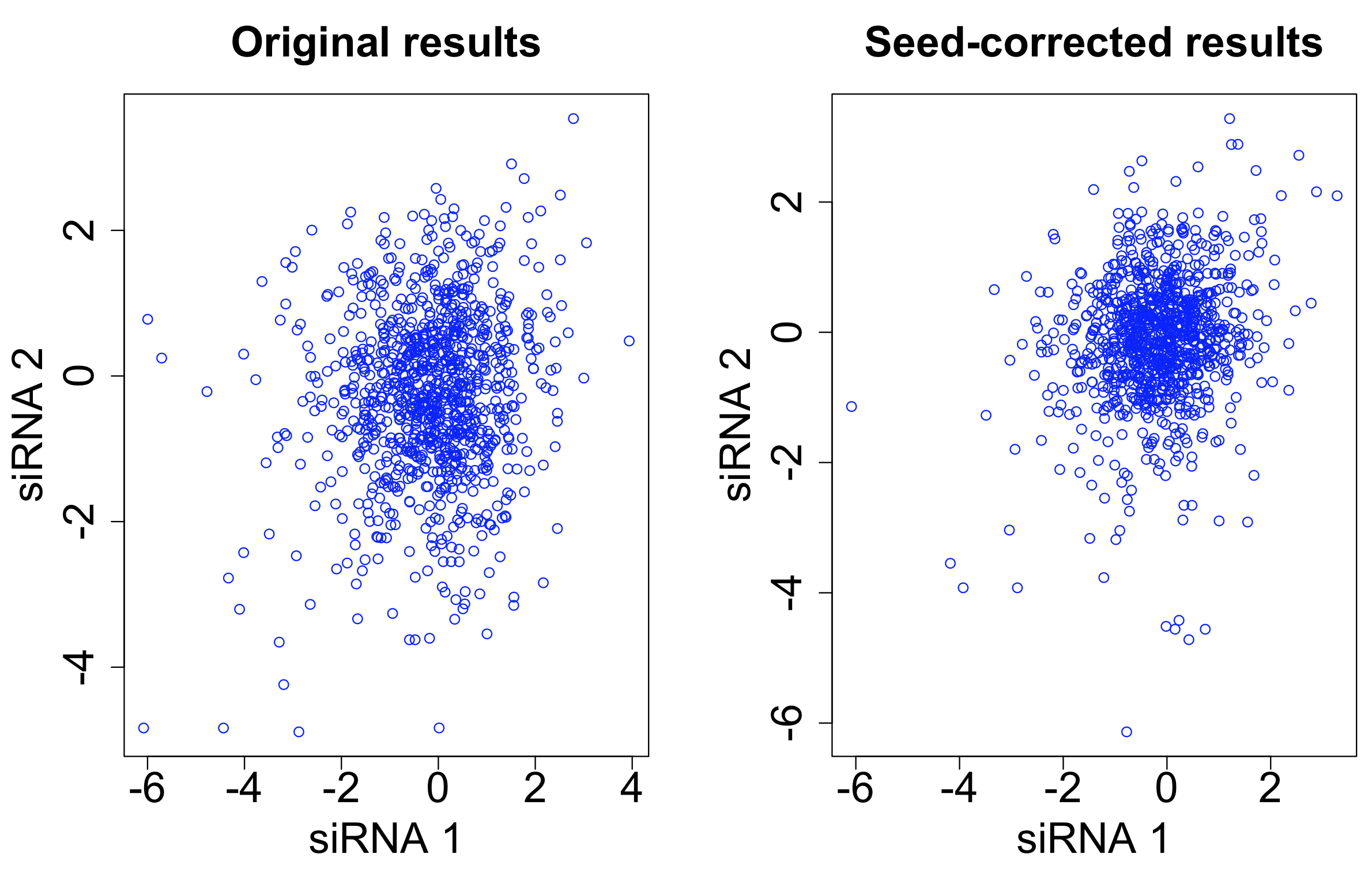
One straightforward way to do seed correction is to subtract the ‘seed median’ from each siRNA. (The seed median is the median for all siRNAs having the given seed.)
This was the approach used by Grohar et al. in a recent genome-wide survey of EWS-FLI1 splicing (involved in Ewing sarcoma). They used the Silencer Select library, which has 3 siRNAs per target gene.
After seed correction, there is only minor improvement in the correlation between siRNAs targeting the same gene. The intra-class correlation (ICC) improves from 0.031 to 0.037. The ICC for siRNAs with the same 7-mer seed decreases from 0.576 to 0.261.
Although we have reduced the seed-based signal, it has not resulted in a correspondingly large improvement in the gene-based signal.
More sophisticated seed correction can improve reagent correlation
Grohar et al. used a simple seed-median subtraction method to correct their screening results.
A more sophisticated method (scsR) was developed by Franceshini et al. for seed-based correction of screening data. It corrects using the mean value for siRNAs with the same seed, and weighs the correction using the standard deviation the values. This allows seeds with a more consistent effect to contribute more to the data normalisation.
Applying the scsR method to the Grohar data, ICC for siRNAs targeting the same gene increases from 0.031 to 0.041. It is better than the increase with seed-median subtraction (0.037), but is still only a fairly minor improvement (plot created using random selection of 10,000 pairs of siRNAs that target the same gene):
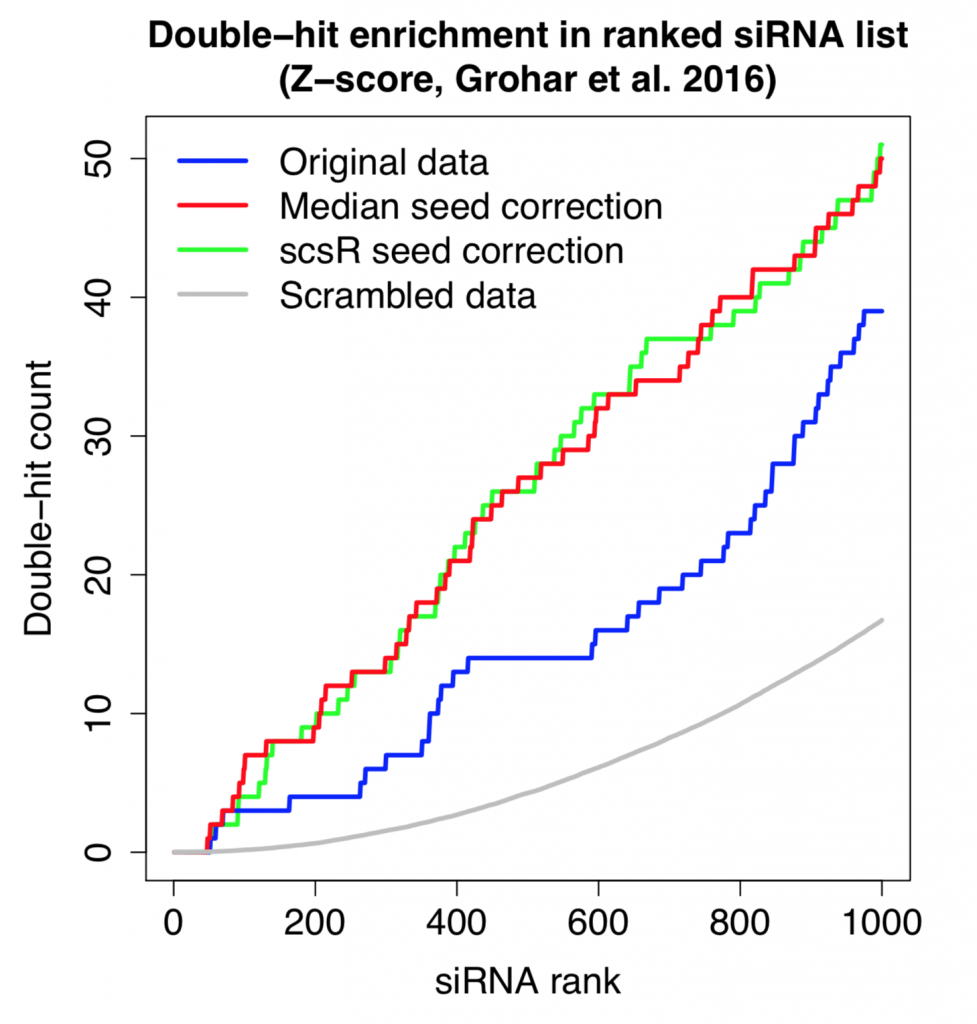
Off-target correction increases double-hit rate in top siRNAs of RNAi screen
The following plot shows the count for single-hit and double-hit genes as we go through the top 1000 siRNAs (of ~60K screened in total). Double-hit means that the gene is covered by 2 (or more) hit siRNAs.
Despite the small improvement in reagent correlation, the double-hit rate is essentially the same using simple seed-median subtraction or the more advanced scsR method.
Furthermore, the number of double-hits is higher than what we’d expect by chance.
This shows that, despite the noise from off-target effects, there is some on-target signal that can be detected.
siRNAs with the strongest phenotypes remain difficult to interpret
Despite the fact that the double-hit count is higher than expected by change, most of the genes targeted by the strongest siRNAs are single-hits. siRNAs with the strongest phenotypes remain difficult to interpret.
Seed correction is best suited for single-siRNA libraries. Low-complexity pools, like siGENOME or ON-TARGETplus, are less amenable to effective seed correction since there are (usually) 4 different seeds per pool. This reduces the effectiveness of seed-based correction, even though seed-based off-target effects remain the primary determinant of observed phenotypes (as discussed here, here , and here).
The best way to correct for seed-based off-targets is to avoid them in the first place. Using more specific reagents, like high-complexity siPOOLs, is the key to generating interpretable RNAi screening results.
For help with seed correction or other RNAi screening data analysis with the Phenovault, contact us at info@sitools.de
As discussed previously, deconvoluted Dharmacon siGENOME pools often give surprising results. (Deconvolution is the process of testing the 4 siRNAs in a pool individually. This is usually done in the validation phase of siRNA screens.)
One way to compare the relative contribution of target gene and off-target effects is to calculate the correlation between reagents having the same target gene or the same seed sequence. One of the first things we do when analysing single siRNA screens is to calculate a robust form of the intraclass correlation (rICC, see discussion at bottom for more about this).
Recently we were analysing deconvolution data from Adamson et al. (2012) and calculated the following rICC’s. (The phenotype measured was relative homologous recombination.)
Besides the order of magnitude difference between target gene and antisense seed correlation (which is commonly observed in RNAi screens), what stands out is the ~2-fold difference between the correlation by target gene and sense seed.
Very little of the the sense strand should be loaded into RISC, if the siRNAs were designed with appropriate thermodynamic considerations (the 5′ antisense end should be less stable than the 5′ sense end, to ensure that the antisense strand is preferentially loaded into RISC).
The above correlations suggest that some not insubstantial amount of sense strand is making it into the RISC complex.
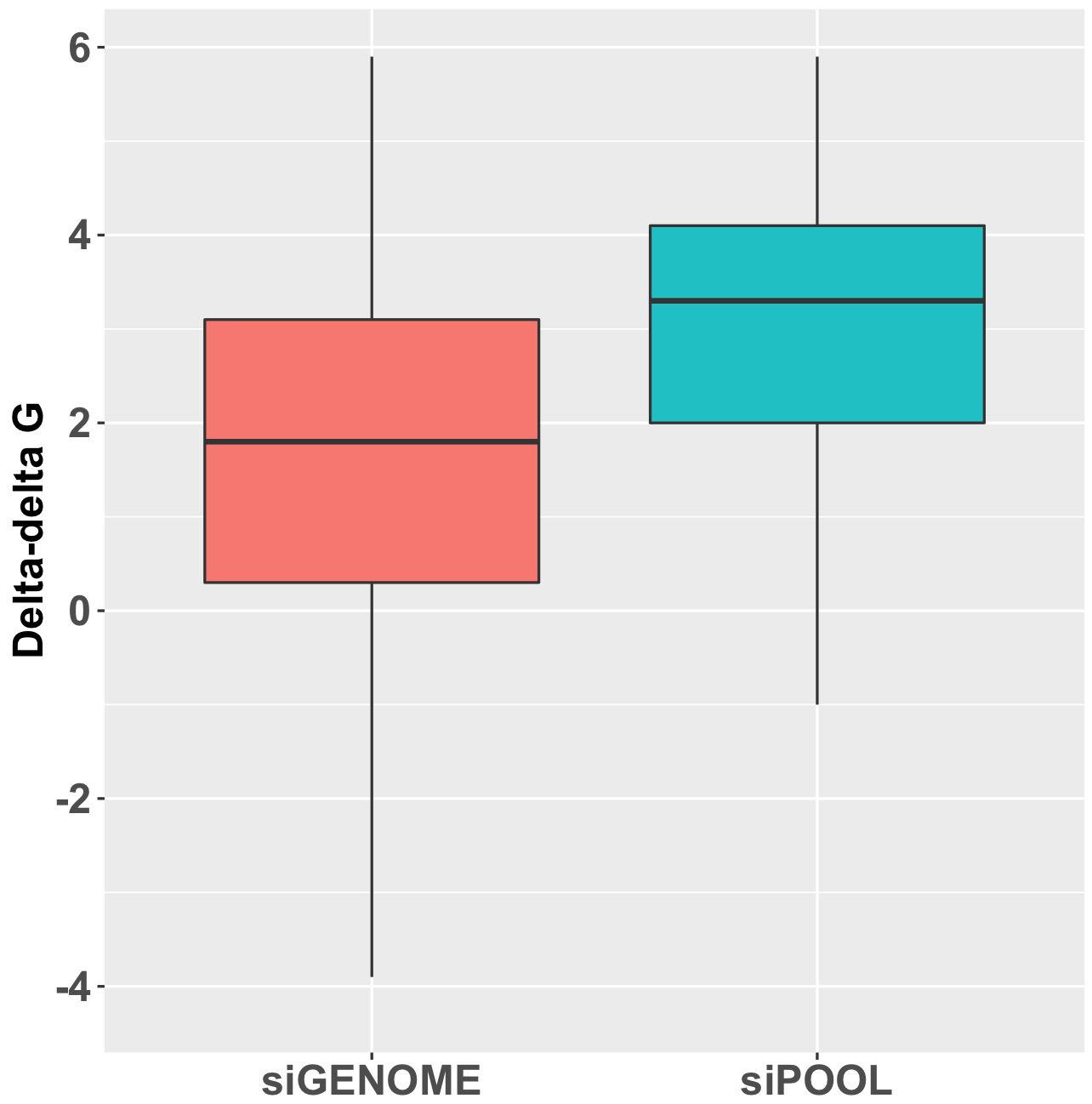
Here is the distribution of delta-delta-G for siPOOLs and siGENOME siRNAs targeting the same 500 human kinases (see bottom of post for discussion of calculation). A positive delta-delta G means that the sense end is more thermodynamically stable than the antisense end, favouring the loading of the antisense strand into RISC.
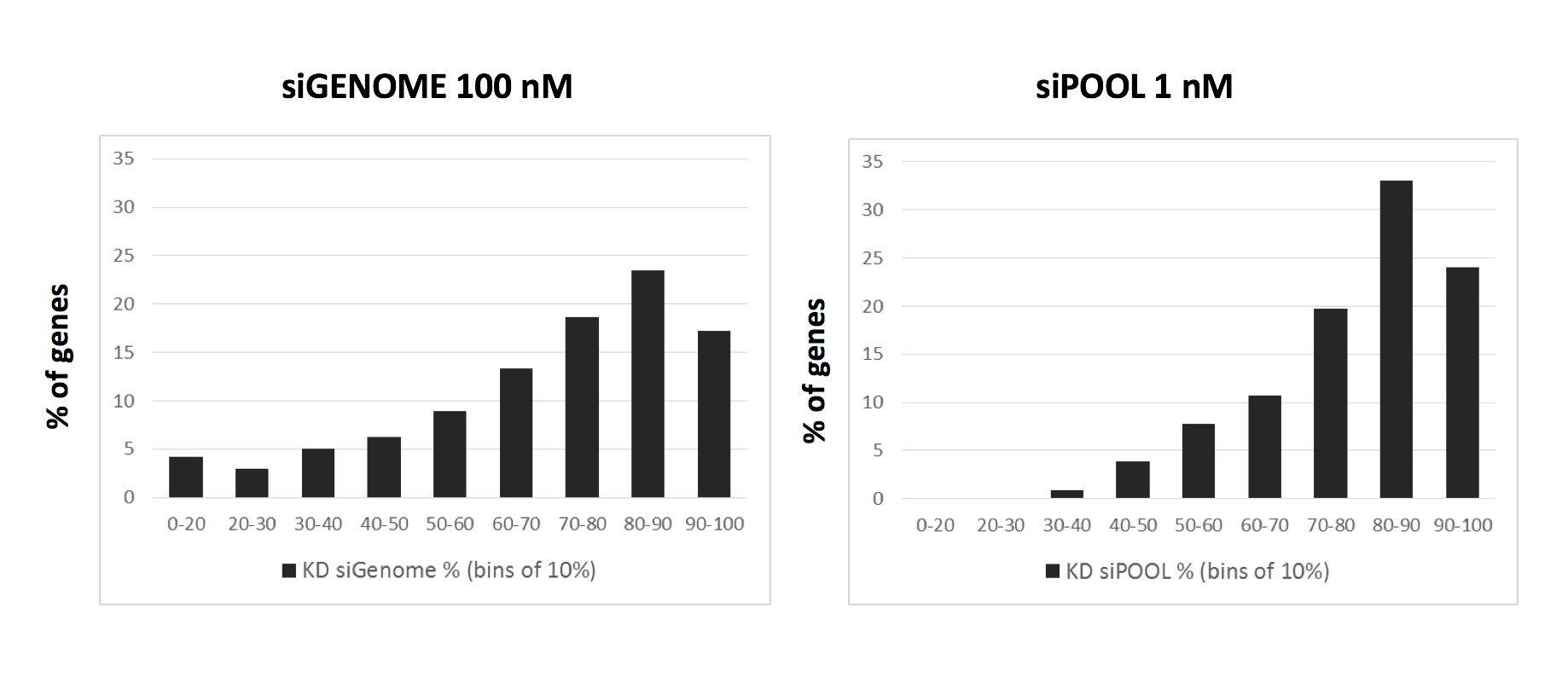
This discrepancy in delta-delta G is also consistent with comparison of mRNA knockdown:
The siGENOME knockdown data comes from 774 genes analysed by qPCR in Simpson et al. (2008). The siPOOL knockdown data is from 223 genes where we have done qPCR validation.
Of note, the siGENOME pools were tested at 100 nM, whereas siPOOLs were tested at 1 nM.
(It should be mentioned that, although consistent with the observed differences in ddG, this is only an indirect comparison, and delta-delta G is not the only determinant of functional siRNAs.)
Notes on intraclass correlation
Intraclass correlation measures the agreement between multiple measurements (in this case, multiple siRNAs with the same target gene, or multiple siRNAs with the same seed sequence). One could also pair off all the repeated measures and calculate correlation using standard methods (parametrically using Pearson’s method, or non-parametrically using Spearman’s method). The main problem with such an approach is that there is no natural way to determine which measure goes in the x or y column. Correlations are normally between different variables (e.g. height and weight). In a case of repeated measures, there is no natural order, so the intraclass correlation (ICC) is the more correct way to measure the similarity of within-group measurements. As ICC depends on a normal distribution, datasets must first be examined, and if necessary, transformed beforehand.
Robust methods have the advantage of permitting the use of untransformed data, which is especially useful when running scripts across hundreds of screening dataset features. The algorithm we use calculates a robust approximation of the ICC by combining resampling and non-parametric correlation.
Here is the algorithm, in a nutshell:
Group observations (e.g. cell count) by the grouping variable (e.g. target gene or antisense seed)
Randomly assign one value of each group to the x or y column (groups with one 1 observation are skipped)
for example, if the grouping variable is target gene and siRNAs targeting PLK1 had the values 23, 30, 37, 45, the program would randomly choose 1 of the values for the x column and another for the y column
Calcule Spearman’s rho (non-parametric measure of correlation)
Repeat steps 1-3 a set number of times (e.g. 300) and store the calculated rho’s
Calculate mean of the rho values from 4. This is the robust approximation of the ICC (rICC).
Values from 4 are also used to calculate confidence intervals.
The program that calculates this is available upon request.
Notes on calculating delta-delta G
Delta-delta G was calculated using the Vienna RNA package, as detailed here: https://www.biostars.org/p/58979/ (in answer by Brad Chapman).
The delta-delta G was calculated using 3 terminal bps. We found that that ddG of the terminal 3 bps had the strongest correlation with observed knockdown. Others (e.g. Schwarz et al., 2003 and Khvorova et al., 2003) have also used the terminal 4 bps.
Want to receive regular blog updates? Sign up for our siTOOLs Newsletter:
Like what you see? Mouse over icons to Follow / Share



 for Ambion vs. Dharmacon siRNAs grouped by same 7mer seed")