A 2013 study on Parkin translocation used genome-wide siRNA libraries from Ambion (single Silencer Select siRNAs) and Dharmacon (pools of 4 siGENOME siRNAs).
The correlation between results for the same on-target gene from the two libraries was very low (R = 0.09). (Each point in the following plot is for a gene.)
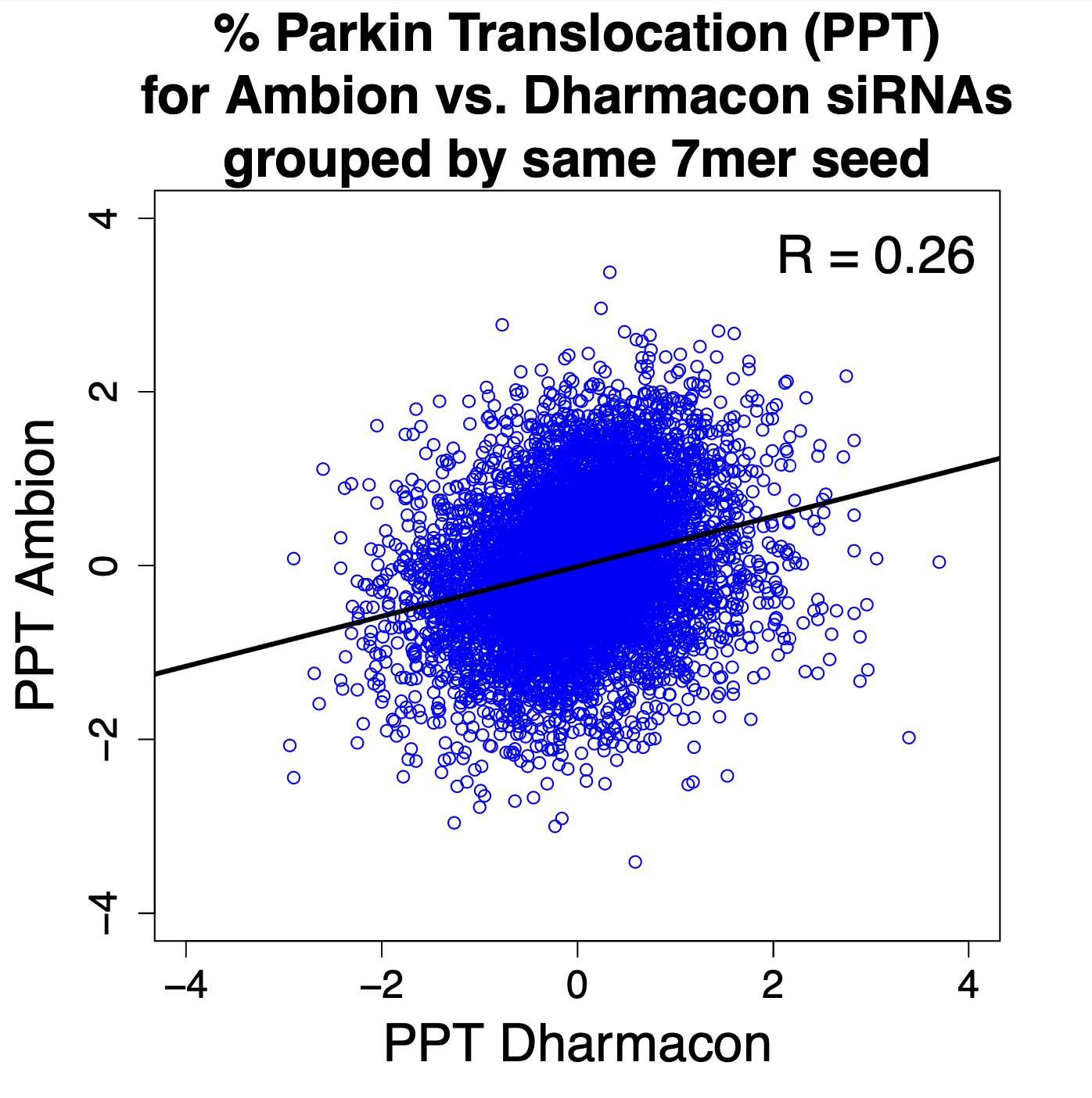
The correlation between results for the same 7mer seed were higher (0.26), providing another example of the Iron Law of RNAi Screening. (Each point in the following plot is for a 7mer seed.)
It is also worth noting that the seed-based correlation would likely have been much higher, had the Dharmacon siRNAs been screened individually (see details below).
Conclusion
The only effective way to avoid off-target effects in RNAi screening is to use high-complexity reagents like siPOOLs, which dilute away off-target effects while maintaining strong on-target silencing.
Analysis details
To calculate the Ambion by-gene value, the mean PPT value was taken for the 3 on-target siRNAs for the gene. (The Dharmacon pooled library only has 1 value per gene, so no further calculation is necessary.)
To calculate the Ambion by-seed value, the mean PPT value was taken for all siRNAs with the 7mer. For Dharmacon, the pool value was assigned to each siRNA, and then siRNAs were grouped by their 7mer seed in order to calculate the seed mean. This means that the Dharmacon siRNA seed value is actually the average from 4 different siRNAs (with different seeds). Had the Dharmacon siRNAs been screened individually, the correlation with Ambion seed results would have been higher.
Low hit validation rate for Dharmacon siGENOME screens
Good experimental design is important when validating hits from RNAi screens. Off-target effects from single siRNAs and low-complexity siRNA pools (e.g. Dharmacon siGENOME) result in high false-positive rates that must be sorted out in validation experiments.
Dharmacon siGENOME pools (SMARTpools) have 4 siRNAs, and the most common form of validation is to test the pool siRNAs individually (deconvolution).
Unfortunately, the results of such deconvolution screening rounds are difficult to interpret.
Rather than deconvoluting the pool, a better approach is to test with independent reagents. Should the phenotype be due to the seed effects of an siRNA in the siGENOME pool, the new designs (with presumably different seed sequences) should not show them. (Note that because they have their own potential complicating off-targets, an even better option would be to use a reagent like siPOOLs that minimises the likelihood of off-target effects).
Independent validation reagents was the approach used by Li et al. in a screen looking for enhancers of antiviral protein ZAP activity.
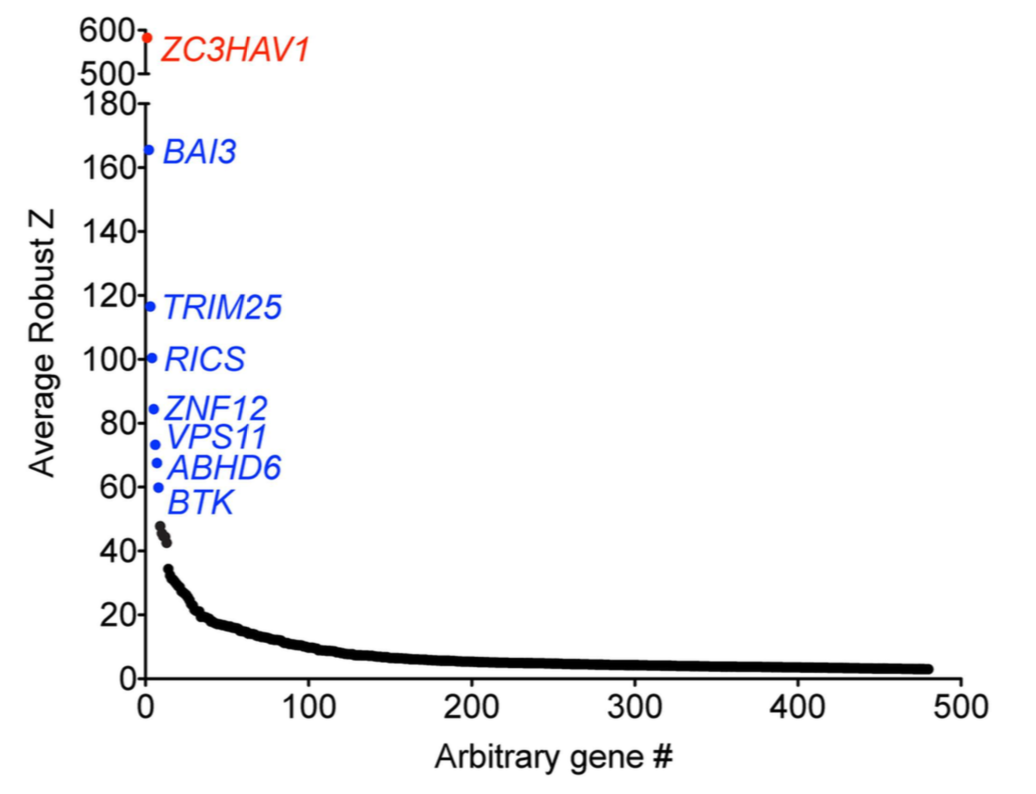
They first did a genome-wide (18,200 genes) screen with siGENOME pools, looking for pools that increased viral infection rate.
The biggest effect was with the positive control, ZAP (aka ZC3HAV1). Several other pools also stood out as giving large increases in viral infection (Fig 1B):
They identified 90 non-control genes with reproducible Z-scores above 3 in their replicate experiments (~0.5% of screened genes).
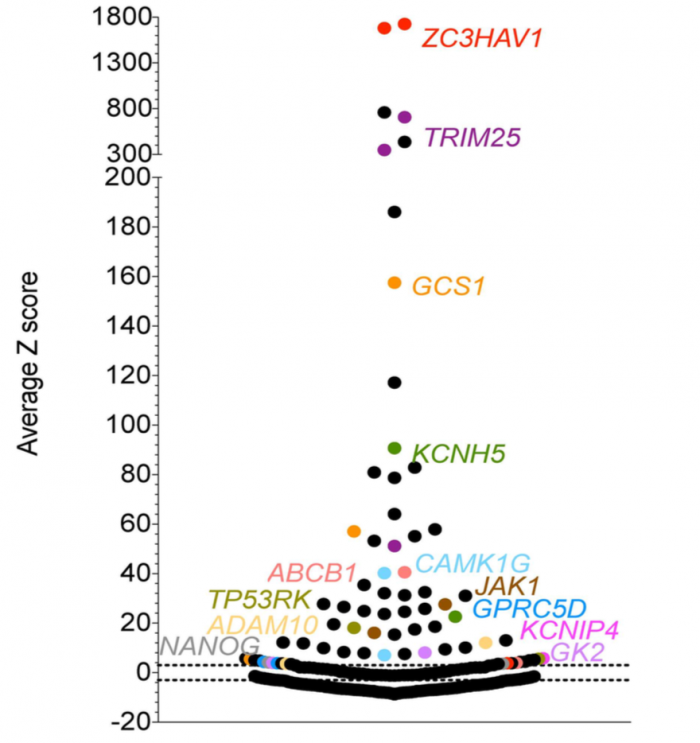
These 90 genes were then tested with 3 Ambion Silencer siRNAs. (They also included a few genes in the validation round based on pathway information and off-target analysis– more on this below.)
Of the 90 candidate hit genes, only 11 could be confirmed (Fig 2B, note that ZC3HAV1/ZAP is the positive control and JAK1 was added to the validation round based on pathway info. A gene was considered confirmed if 2 of 3 siRNAs had a Z-score > 3.):
We also see that only 1 of the 7 top hits from the first round (blue genes in the first figure) was confirmed. This is a common observation in RNAi screens: the strongest phenotypes are mostly due to off-target effects.
Off-target effects are difficult to interpret, even using advanced analysis programs like Haystack or GESS. The authors tested 4 genes identified by Haystack as targets for seed-based off-targeting. None of those genes could be confirmed in the validation round.
Pooling only 4 siRNAs increases off-target effects
In a previous post, we showed how siRNA pools with small numbers of siRNAs can exacerbate off-target effects.
Low-complexity pools (with 4 siRNAs per gene) should thus lead to overall stronger off-target effects than single siRNAs.
This phenomenon was addressed in a bioinformatics paper a few years back. The authors created a model to predict gene phenotypes based on the combined on-target and off-target effects of siRNAs.
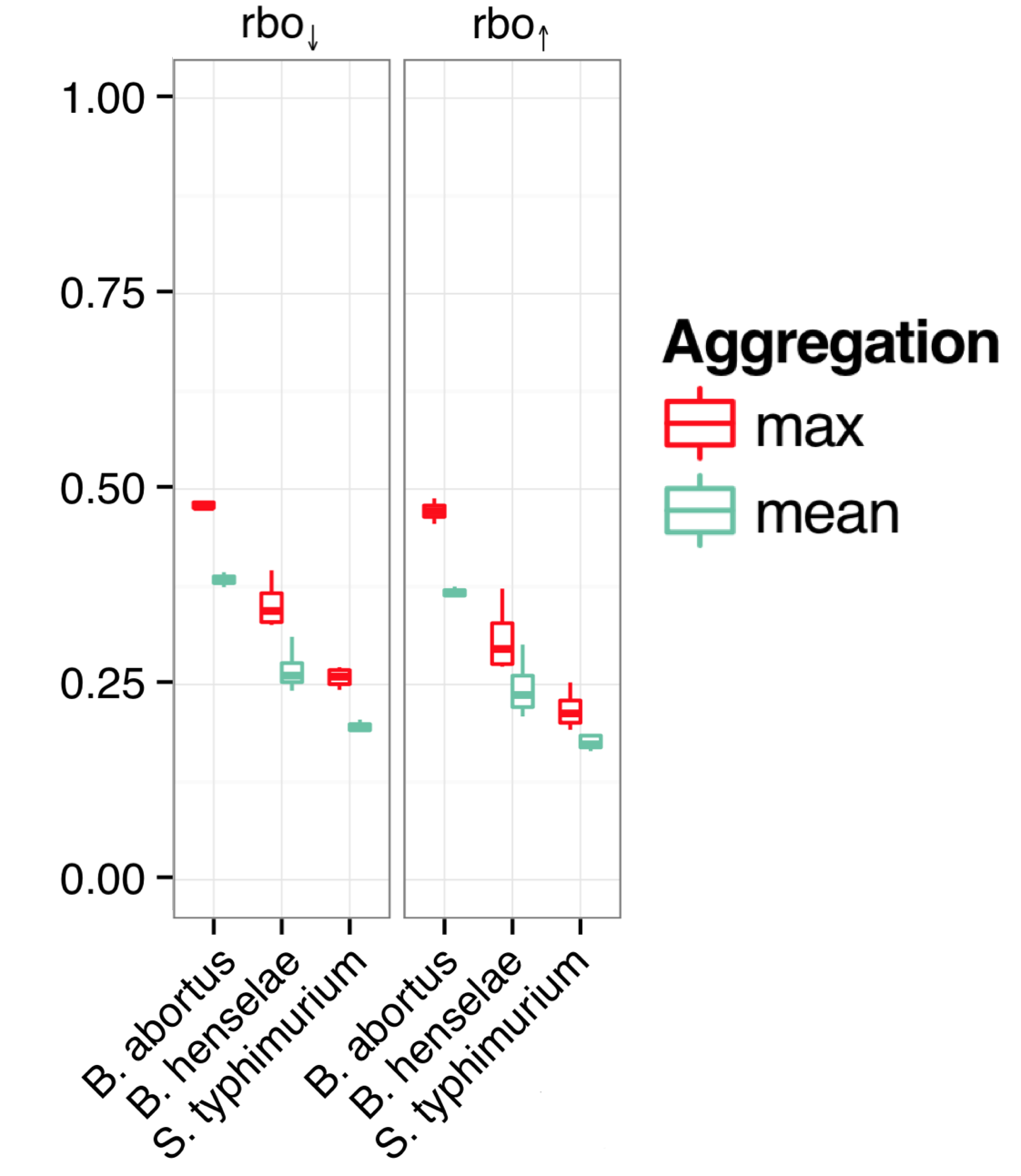
The siRNAs were screened either individually (Ambion and Qiagen), or in pools of four (Dharmacon siGENOME), in 3 different batcterial-infection assays (B. abortus, B. henselae, and S. typhimurium).
The model assumed that each siRNA silenced its on-target gene to the same level. For off-target silencing, they used the predictions from TargetScan, a program for calculating seed-based knockdown by miRNAs or siRNAs.
In order to assess model quality, they checked how similar the gene phenotype predictions were when using different reagents types in the same pathogen-infection screen.
The following figure shows the rank-biased overlap (a measure of how similar lists are with regards to top- and bottom-ranked items), when estimating siGENOME off-target knockdown in one of 2 ways:
A) using the maximum TargetScan score for any of the 4 siRNAs in the siGENOME pool
B) using the mean TargetScan score for the 4 siRNAs
If low-complexity pooling increases the degree of off-target effects, we would expect the maximum TargetScan score to produce better model concordance.
And that is what the authors found. (the two plots show the rank-biased overlap for the top and bottom of the phenotype ranked lists, respectively)
The off-target effect of a 4-siRNA, low-complexity pool is best described by the strongest off-target effect of any of the individual siRNAs.
As discussed in our NAR paper, pooling a minimum of 15 siRNAs is required to reliably prevent off-target effects.
This blogpost describes issues encountered in target validation and how to safeguard against poor reproducibility in RNAi experiments.
The importance of target validation
More than half of all clinical trials fail from a lack of drug efficacy. One of the major reasons for this is inadequate target validation.
Target validation involves verifying whether a target (protein/nucleic acid) merits the development of a drug (small molecule/biologic) for therapeutic application.
Failing to adequately validate a target can burden a pharma with roughly 800 million to 1.4 billion in drug development costs. Impact is not only monetary as largesite closures often result as companies struggle to save costs and a reduced production effort deprives patients of new medicines.
Performing target validation well
Special attention should therefore be given to performing target validation techniques well.
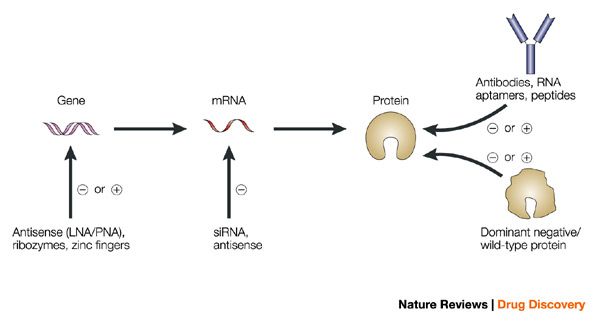
Overview of target validation techniques (Lindsay, Nat Review Drug Discovery, 2013)
Many of these techniques involve inhibiting target expression to establish its relevance in a cellular or animal disease model. This can be performed with chemical probes, RNA interference (RNAi), genetic knock-outs, and even targeted protein degradation.
The reproducibility of these techniques however has been an issue of concern for drug developers. Less than half of all findings from peer-reviewed scientific publications was reported to be successfully reproduced.
Dismal rates of reproducibility from several pharma-led cancer-focused studies ranged from 11% (Amgen) to 25% (Bayer). A review by William Kaelin Jr sums up the common pitfalls of preclinical cancer target validation. One of his key points:
Cellular phenotypes caused by a chemical or genetic perturbant should be considered to be off-target until proved otherwise, especially when the phenotypes were detected in a down assay and therefore could reflect a nonspecific loss of cellular fitness. It is only by performing rescue experiments that one can formally address whether the effects of a perturbant are on-target.
The comment highlights the issue of reagent non-specificity as a notable contribution towards poor reproducibility.
Certainly, for RNAi the wide-spread off-target effects of siRNAs has been observed in numerous publications. The mechanism being well-established to be based on microRNA-like seed-based recognition of non-target genes. The effect dominates over on-target effects in many large RNAi screens, illustrating the depth of the problem.
Reagent non-specificity is not restricted to RNAi. There have been multiple reports of non-specificity for gene editing technique, CRISPR, which can be read about in detail here, here and here. Recent publications continue to shed more light on its potential off-targets as we learn more about this relatively new technique.
Even chemical probes may have multiple targets. It is hence imperative that more than one target validation technique be used to avoid confirmation bias.
Target validation – a story from Pharma
Back in 2013, when siTOOLs was just starting out, a pharma approached us with a target validation problem.
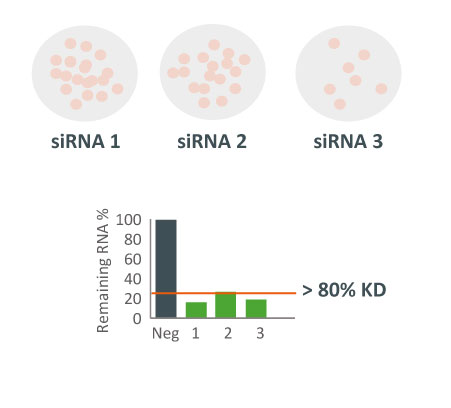
They were obtaining different results with 3 different siRNAs in a cellular proliferation assay. Despite all 3 siRNAs potently downregulating the target gene, they produced different effects on cell viability.
Which siRNA tool to trust?
Three different siRNAs against the same target were tested in a cell proliferation assay. Despite all 3 siRNAs showing potent target gene silencing, effect on cell proliferation differed greatly.
A whole-transcriptome expression analysis performed for the 3 siRNAs and a siPOOL designed against the same target revealed the reason for the large variability.
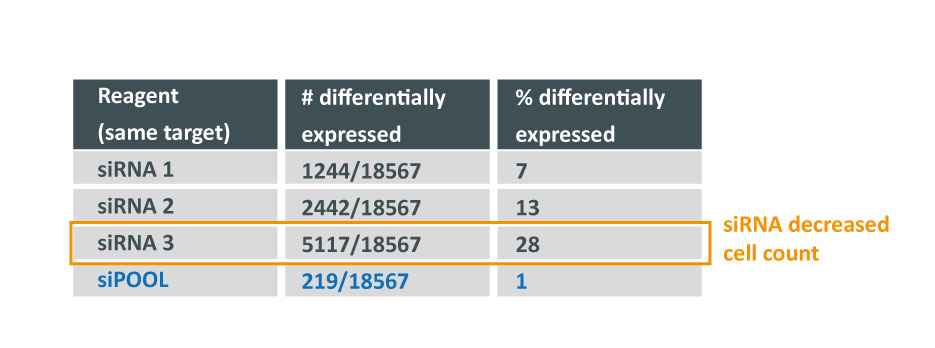
How many genes can you affect with an siRNA? Whole transcriptome analysis by microarray was performed and number and % of up and down-regulated genes are shown over total number of genes assayed (18567).
Despite all siRNA tools affecting the same target, the difference in extent of gene deregulation was astounding. With the greatest number of off-target effects, it was not surprising that siRNA 3 showed an impact on cell proliferation.
In contrast, siPOOLs had 5 to 25X less differentially expressed genes compared to the 3 commercial siRNAs against the same target. An expression analysis carried out for another gene target showed similar results i.e. siPOOLs having far less off-targets.
The target was dropped from development. A great example where failing early is a good thing, though it was not without costs from validating the multiple siRNAs.
The recommended target validation tool
Functioning like a pack of wolves, siPOOLs increase the chances of capturing large and difficult prey, while making full use of group diversity to compensate for individual weakness.
siPOOLs efficiently counter RNAi off-target effects by high complexity pooling of sequence-defined siRNAs. This enables individual siRNAs to be administered at much lower concentrations, below the threshold for stimulating significant off-target gene deregulation. Due to having multiple siRNAs against the same target gene, target gene knock-down is maintained and in fact becomes more efficient.
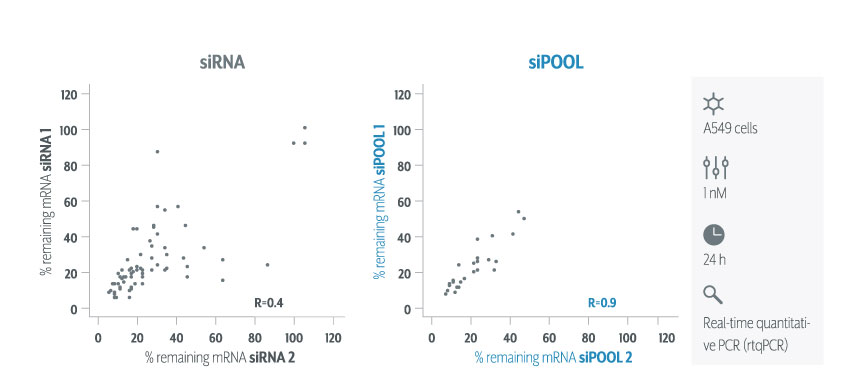
siPOOLs increase targeting efficiency, avoiding knock-down variability. Figure shows rtqPCR quantification of target RNA levels when two siPOOLs vs two siRNAs against 36 genes were tested.
We still recommend using multiple target validation techniques. As a first evaluation however, siPOOLs are quick, easy and most of all, reliable.
Summary:Low-complexity siRNA pooling (e.g. Dharmacon siGENOME SMARTpools) does not prevent siRNA off-targets. It may in fact exacerbate off-target effects. Only high-complexity pooling (siPOOLs) can reliably ensure on-target phenotypes.
Low-complexity pooling increases the number of siRNA off-targets
One of the claims often made in favour of low-complexity pooling (e.g Dharmacon siGENOME SMARTpools) is that this pooling reduces the number of seed-based off-target effects compared to single siRNAs.
If this were true, we would expect different low-complexity siRNA pools for the same gene to give similar phenotypes. But this is not the case.
Published expression data shows that low-complexity pooling actually increases the number of off-targets.
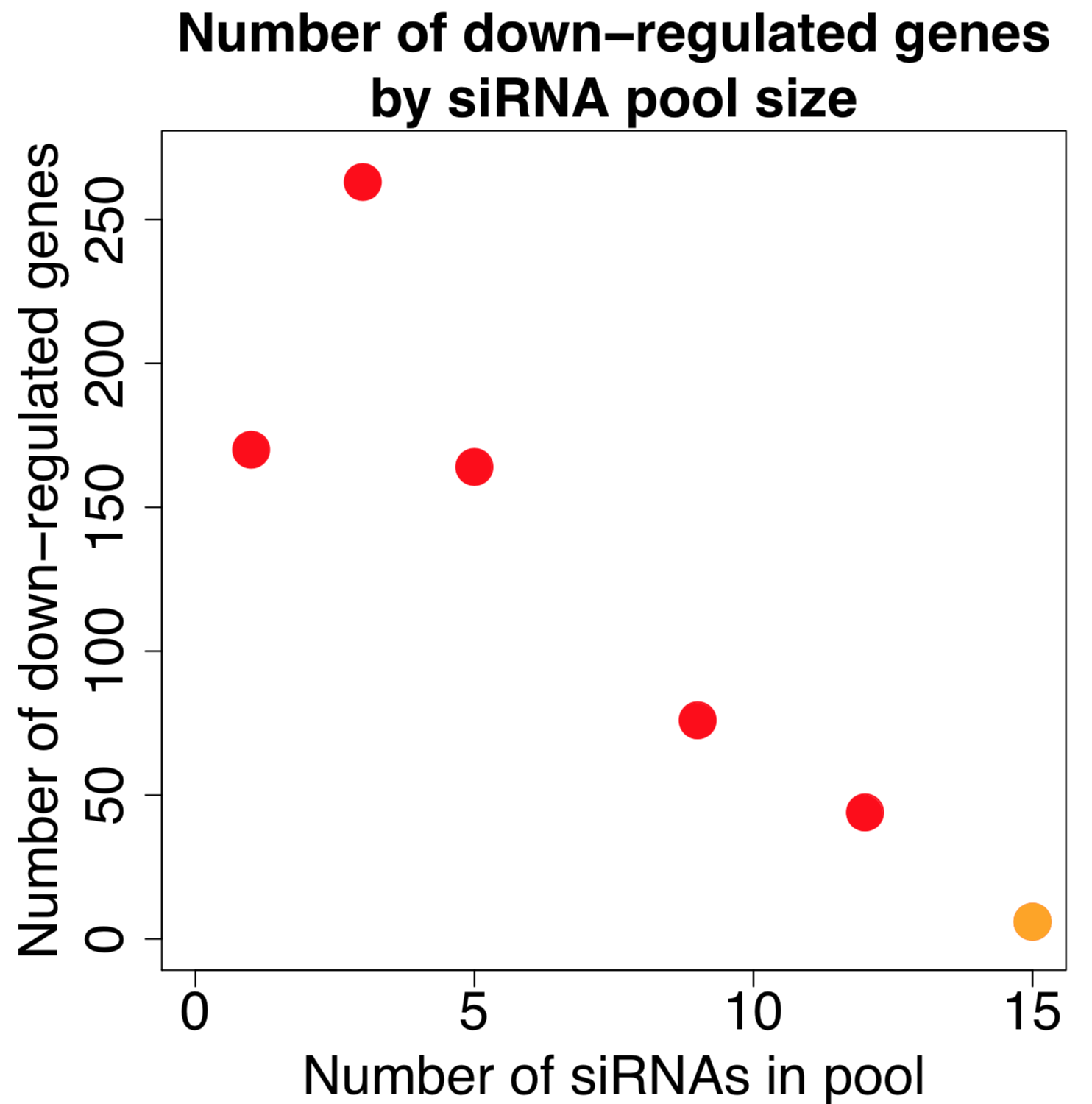
Kittler et al. (2007) looked at the effect of combining differing number of siRNAs in low to medium complexity siRNA pools (siRNA pools sizes were: 1, 3, 5, 9, and 12).
Their work showed that the number of down-regulated genes (50% or greater silencing) actually increases when small numbers of siRNAs are combined. Only when larger numbers of siRNAs are combined does the number of off-targets start to drop:
[The figure is based on data from GEO dataset GSE6807. Down-regulated genes are those whose expression is reduced by 50% or more. Note that the orange point is taken from our 2014 NAR paper, as we are not aware of other published expression datasets with this many pooled siRNAs. A few caveats with combining these datasets are that they use different target genes, siRNA concentrations, and the data comes from a different expression platform.]
Low-complexity pooling: a bad solution for siRNA off-targets
Low-complexity pooling does not get rid of the main problem associated with single siRNAs: seed-based off-target effects. Based the above analysis, it can make it even worse. It also prevents use of the most effective computational measures against seed effects.
Redundant siRNA Activity (RSA) is a common on-target hit analysis method for single-siRNA screens. It checks how over-represented the siRNAs for a gene are at the top of a ranked screening list. If a gene has 2 or more siRNAs near the top of the list, it will score better than a gene that only has a single siRNA near the top of the list. This is one way to reduce the influence of strong off-target siRNAs.
Correcting single siRNA values by seed medians has also been shown to be an effective way to increase the on-target signal in screens. This correction is not effective for low-complexity pools, since each pool can contain 3-4 different seeds.
Off-target based hit detection algorithms (e.g. Haystack and GESS) are also only effective for single-siRNA screens. The advantage of these algorithms is that it permits the detection of hit genes that were not screened with on-target siRNAs. These algorithms are not effective for low-complexity pool screens.
Our recommendation: do not convert single siRNAs into low-complexity pools, rather use high-complexity siPOOLs to confirm hits
We do not recommend that screeners combine their single siRNA libraries into low-complexity pools (e.g. combining 3 Silencer Select siRNAs for the same target gene). If possible, it is better to screen the siRNAs individually and then apply seed-based correction, RSA and seed-based hit-detection algorithms.
The time saved by only screening one well per target may prove illusory when the deconvolution experiments show that the individual siRNAs have divergent phenotypes.
It is probably better to deal with off-target effects up front (by screening single siRNAs) than to be surprised by them later in the screen (during pool deconvolution).
Summary: Conventional siRNAs have a high probability of giving off-target phenotypes. siRNA off-target effects can be reduced by using more specific reagents or narrowing the assay focus (to reduce the number of relevant genes). Even when the assay is relatively focused, more specific reagents significantly increase the probability of observing on-target effects.
Probability of siRNA off-target phenotype depends on reagent specificity and assay biology
The probability of getting an off-target effect from an siRNA depends on several factors, the main ones being reagent specificity and assay biology. If an siRNA down-regulates a large number of genes, or if an assay phenotype can be induced by a large number of genes, the probability of observing an off-target phenotype increases.
siRNAs can down-regulate many off-target genes
Garcia et al. (2011) compiled 164 different microarray experiments measuring gene expression following transfection with siRNAs. The mean number of down-regulated genes in these experiments was 132 and the median was 68 (down-regulated genes were silenced by 50% or more).
As noted in earlier studies of gene expression following siRNA treatment (e.g. Jackson et al. 2003), few of the down-regulated genes are shared between siRNAs with the same target gene. This suggests that the down-regulated genes are not the downstream result of target gene knockdown (i.e. they are mostly off-target).
High-complexity pooling of siRNAs (e.g. with siPOOLs) can reduce the number of down-regulated genes.
The following figure, based on data from Hannus et al. 2014, shows the difference between the gene expression changes caused by a single siRNA (left) and a high-complexity siRNA pool (siPOOL, right), which also includes that same single siRNA:
Estimating the probability of siRNA off-target phenotypes
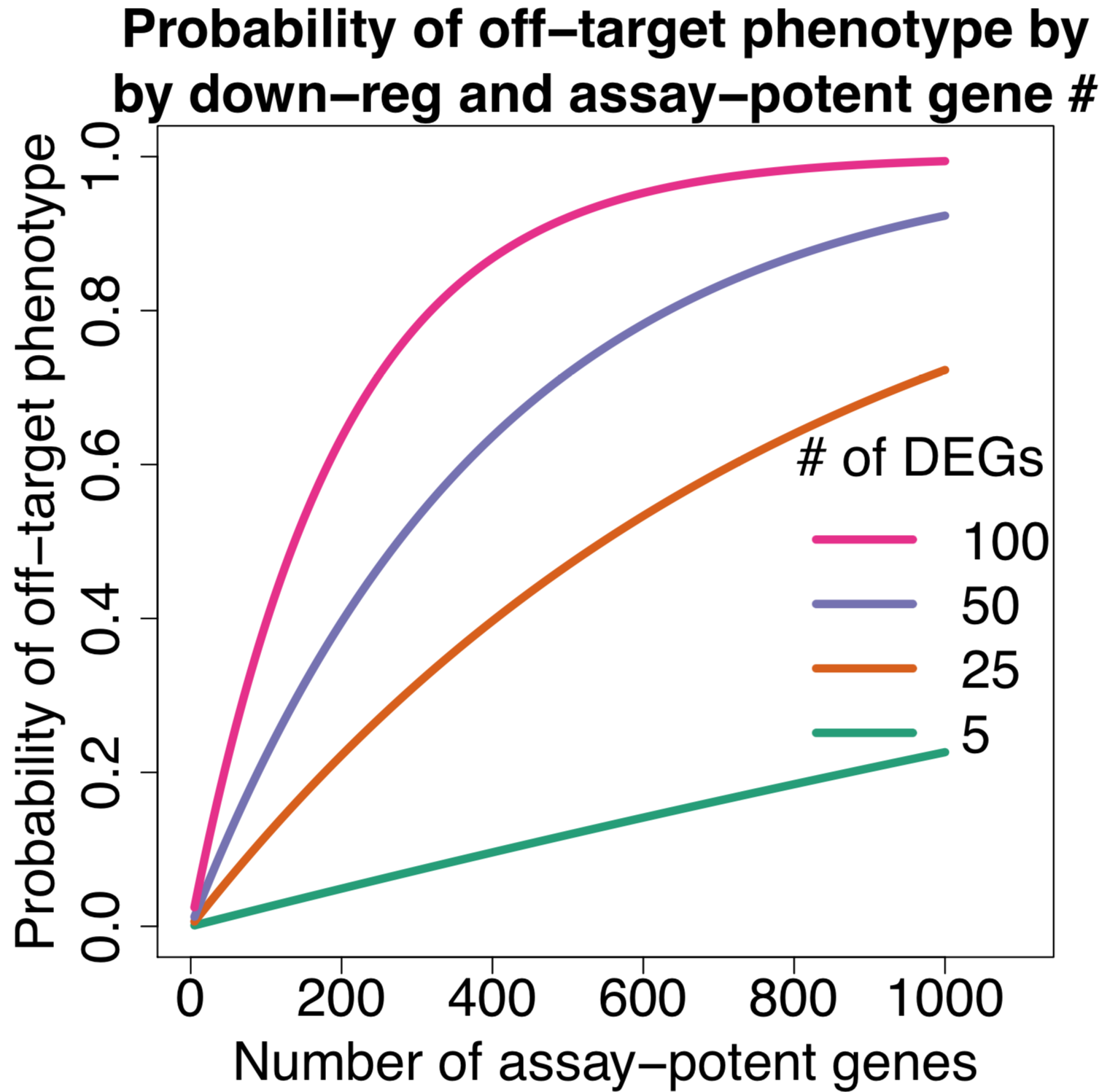
Assuming different numbers of down-regulated genes (off-target) and different numbers of potent genes involved in assay pathways, we can try to estimate the probability of an siRNA giving an off-target effect.
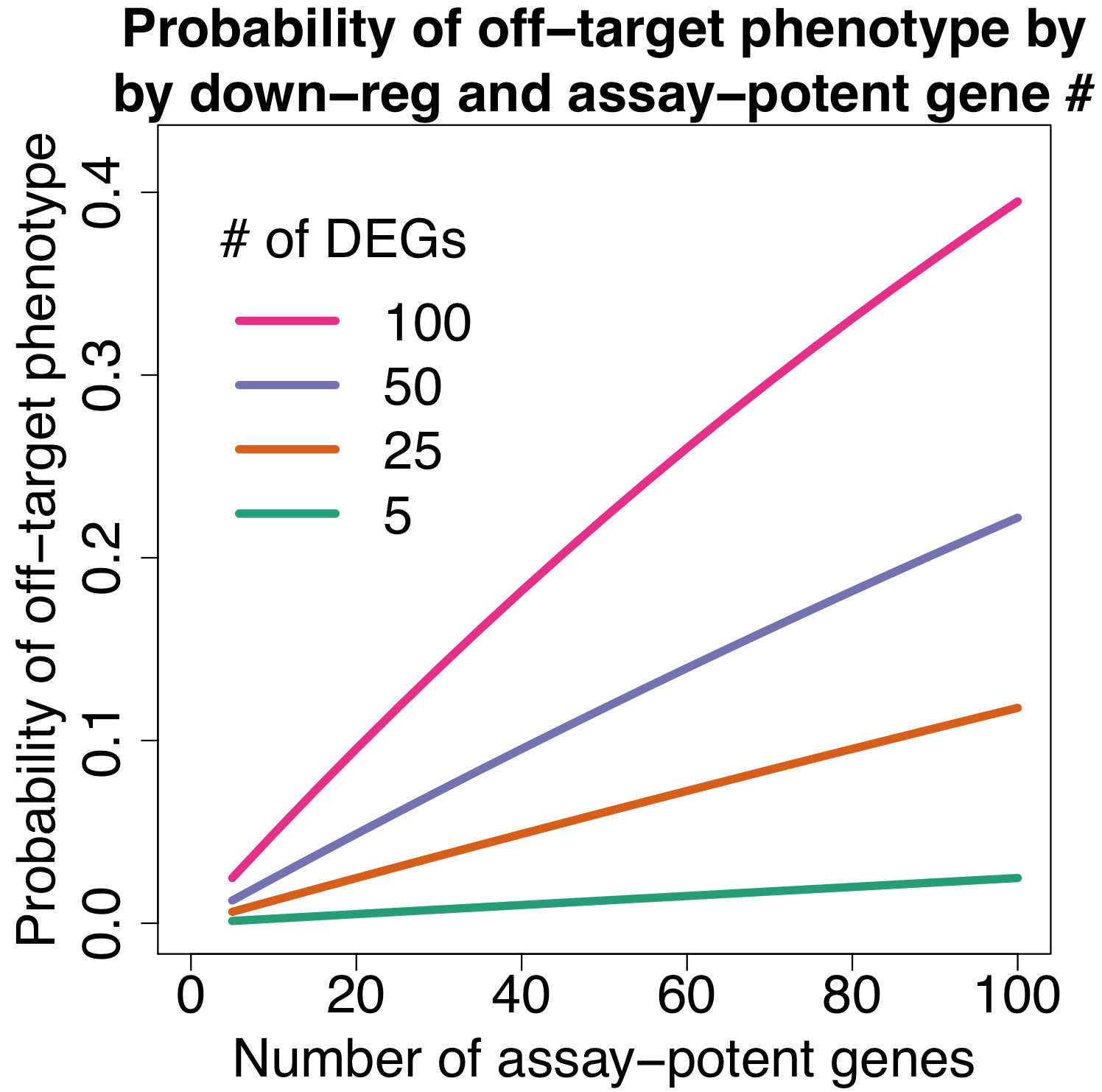
The following plot shows the probability of getting an off-target effect when:
down-regulated means that gene expression is reduced by 50% or more
in the Garcia paper dataset, the mean is 132 and median is 68
assuming different numbers of assay-potent genes
an assay-potent gene is one whose down-regulation by 50% or more is sufficient to produce a hit phenotype
for assays with more general phenotypes (e.g. cell count) we would expect more assay-potent genes
We can see that even if there are only 20 assay-potent genes, there’s a nearly 10% chance of getting an off-target phenotype when siRNAs down-regulate 100 off-target genes (which is close to the average observed in the Garcia dataset).
In a genome-wide screen of 20,000 genes with 3 siRNAs per gene, we would thus expect 2,000 off-target siRNAs.
In contrast, a more specific reagent that only down-regulates 5 off-target genes only has a 0.5% change of producing an off-target phenotype. For the above-mentioned genome-wide RNAi screen, we would expect only 100 off-target siRNAs (a 20-fold reduction).
The importance of RNAi reagent specificity
The above analysis demonstrates the importance of using specific siRNA reagents.
Changing an assay to make the phenotypic readout narrower (to reduce the number of genes capable of inducing a phenotype) is one way to reduce the risk of off-target phenotypes. But this may be a lot of work and is not necessarily desirable or even possible.
A more ideal solution is the use of a specific RNAi reagent, like siPOOLs.
postscript
As the number of assay-potent genes increases, the probability of getting an off-target phenotype approaches one.
The following plot (same format as the one above) shows the distribution
The p-values were calculated using the hypergeometric distribution, assuming a population size of 20,000 (the approximate number of protein-coding genes in the human genome).
Note that one of the major simplifying assumptions of the above analysis is that all siRNAs have the same number of down-regulated off-target genes.
Correcting seed-based off-target effects in RNAi screens
Summary: Correcting for seed-based off-targets can improve the results from RNAi screening. However, the correlation between siRNAs for the same gene is still poor and the strongest screening hits remain difficult to interpret.
Seed-based off-target correction has little effect on reagent reproducibility
Given that seed-based off-targets are the main cause of phenotypes in RNAi screening, trying to correct for those effects makes good sense.
The dominance of seed-based off-targets means that independent siRNAs for the same gene usually show poor correlation.
If one could correct for the seed effect, the correlation between siRNAs targeting the same gene may improve.
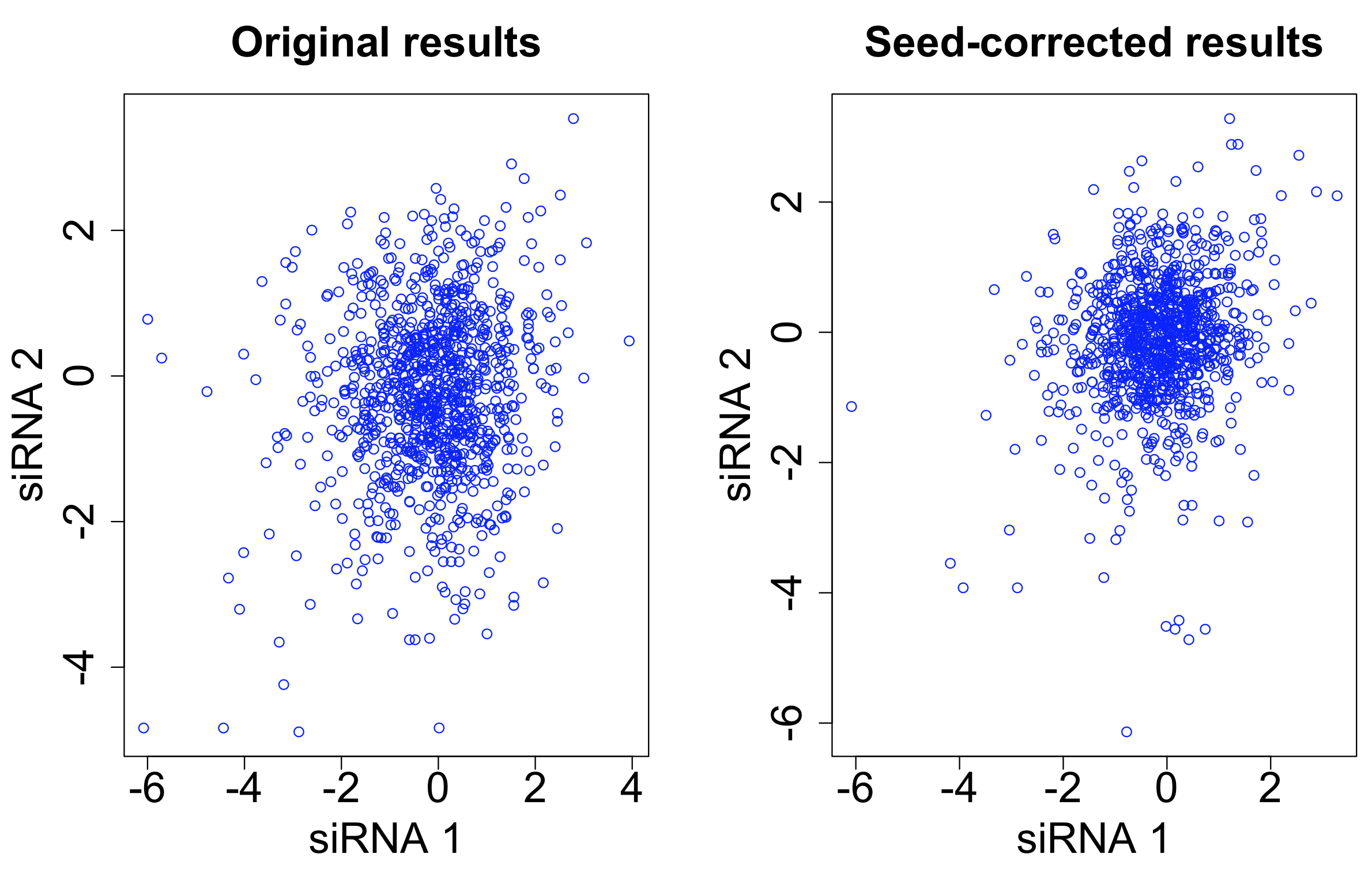
One straightforward way to do seed correction is to subtract the ‘seed median’ from each siRNA. (The seed median is the median for all siRNAs having the given seed.)
This was the approach used by Grohar et al. in a recent genome-wide survey of EWS-FLI1 splicing (involved in Ewing sarcoma). They used the Silencer Select library, which has 3 siRNAs per target gene.
After seed correction, there is only minor improvement in the correlation between siRNAs targeting the same gene. The intra-class correlation (ICC) improves from 0.031 to 0.037. The ICC for siRNAs with the same 7-mer seed decreases from 0.576 to 0.261.
Although we have reduced the seed-based signal, it has not resulted in a correspondingly large improvement in the gene-based signal.
More sophisticated seed correction can improve reagent correlation
Grohar et al. used a simple seed-median subtraction method to correct their screening results.
A more sophisticated method (scsR) was developed by Franceshini et al. for seed-based correction of screening data. It corrects using the mean value for siRNAs with the same seed, and weighs the correction using the standard deviation the values. This allows seeds with a more consistent effect to contribute more to the data normalisation.
Applying the scsR method to the Grohar data, ICC for siRNAs targeting the same gene increases from 0.031 to 0.041. It is better than the increase with seed-median subtraction (0.037), but is still only a fairly minor improvement (plot created using random selection of 10,000 pairs of siRNAs that target the same gene):
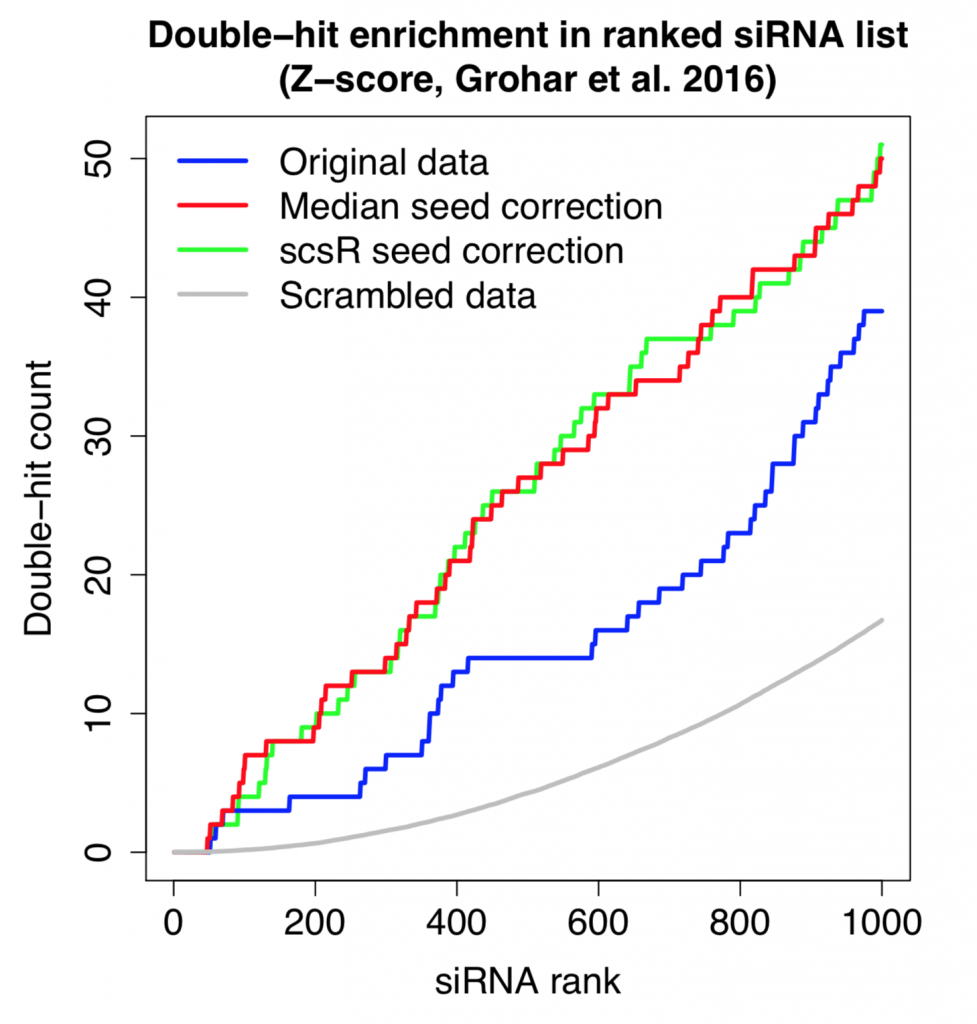
Off-target correction increases double-hit rate in top siRNAs of RNAi screen
The following plot shows the count for single-hit and double-hit genes as we go through the top 1000 siRNAs (of ~60K screened in total). Double-hit means that the gene is covered by 2 (or more) hit siRNAs.
Despite the small improvement in reagent correlation, the double-hit rate is essentially the same using simple seed-median subtraction or the more advanced scsR method.
Furthermore, the number of double-hits is higher than what we’d expect by chance.
This shows that, despite the noise from off-target effects, there is some on-target signal that can be detected.
siRNAs with the strongest phenotypes remain difficult to interpret
Despite the fact that the double-hit count is higher than expected by change, most of the genes targeted by the strongest siRNAs are single-hits. siRNAs with the strongest phenotypes remain difficult to interpret.
Seed correction is best suited for single-siRNA libraries. Low-complexity pools, like siGENOME or ON-TARGETplus, are less amenable to effective seed correction since there are (usually) 4 different seeds per pool. This reduces the effectiveness of seed-based correction, even though seed-based off-target effects remain the primary determinant of observed phenotypes (as discussed here, here , and here).
The best way to correct for seed-based off-targets is to avoid them in the first place. Using more specific reagents, like high-complexity siPOOLs, is the key to generating interpretable RNAi screening results.
For help with seed correction or other RNAi screening data analysis with the Phenovault, contact us at info@sitools.de
As discussed previously, deconvoluted Dharmacon siGENOME pools often give surprising results. (Deconvolution is the process of testing the 4 siRNAs in a pool individually. This is usually done in the validation phase of siRNA screens.)
One way to compare the relative contribution of target gene and off-target effects is to calculate the correlation between reagents having the same target gene or the same seed sequence. One of the first things we do when analysing single siRNA screens is to calculate a robust form of the intraclass correlation (rICC, see discussion at bottom for more about this).
Recently we were analysing deconvolution data from Adamson et al. (2012) and calculated the following rICC’s. (The phenotype measured was relative homologous recombination.)
Besides the order of magnitude difference between target gene and antisense seed correlation (which is commonly observed in RNAi screens), what stands out is the ~2-fold difference between the correlation by target gene and sense seed.
Very little of the the sense strand should be loaded into RISC, if the siRNAs were designed with appropriate thermodynamic considerations (the 5′ antisense end should be less stable than the 5′ sense end, to ensure that the antisense strand is preferentially loaded into RISC).
The above correlations suggest that some not insubstantial amount of sense strand is making it into the RISC complex.
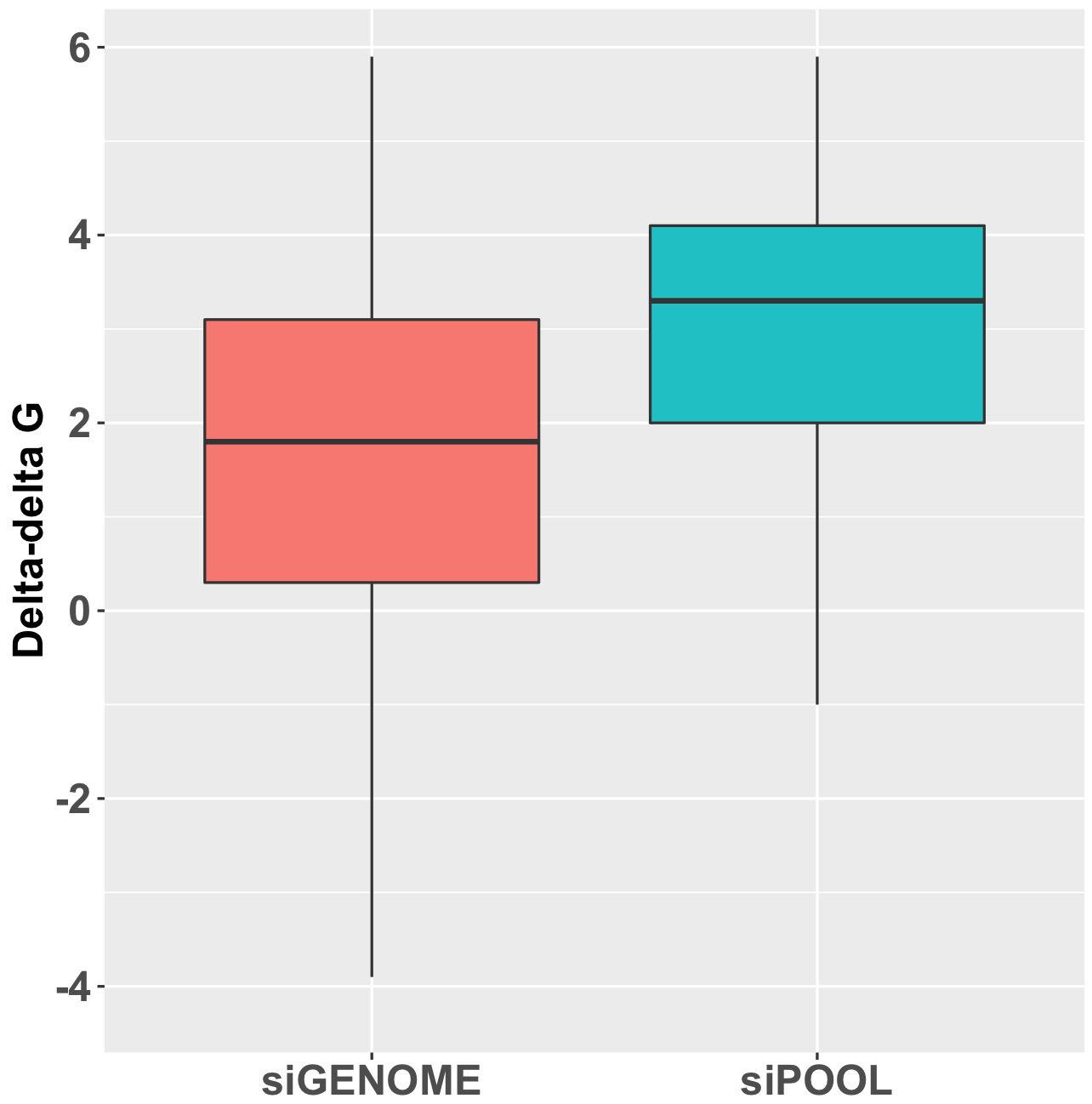
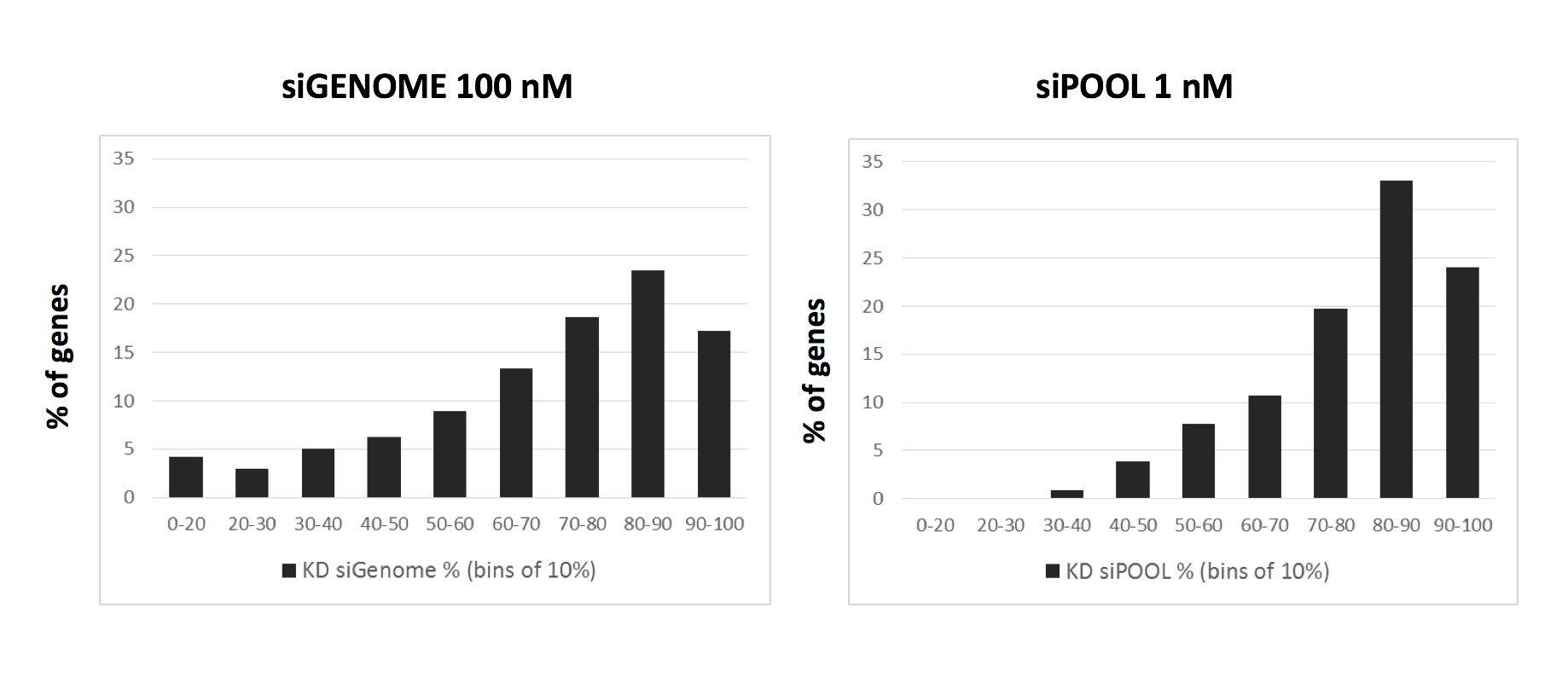
Here is the distribution of delta-delta-G for siPOOLs and siGENOME siRNAs targeting the same 500 human kinases (see bottom of post for discussion of calculation). A positive delta-delta G means that the sense end is more thermodynamically stable than the antisense end, favouring the loading of the antisense strand into RISC.
This discrepancy in delta-delta G is also consistent with comparison of mRNA knockdown:
The siGENOME knockdown data comes from 774 genes analysed by qPCR in Simpson et al. (2008). The siPOOL knockdown data is from 223 genes where we have done qPCR validation.
Of note, the siGENOME pools were tested at 100 nM, whereas siPOOLs were tested at 1 nM.
(It should be mentioned that, although consistent with the observed differences in ddG, this is only an indirect comparison, and delta-delta G is not the only determinant of functional siRNAs.)
Notes on intraclass correlation
Intraclass correlation measures the agreement between multiple measurements (in this case, multiple siRNAs with the same target gene, or multiple siRNAs with the same seed sequence). One could also pair off all the repeated measures and calculate correlation using standard methods (parametrically using Pearson’s method, or non-parametrically using Spearman’s method). The main problem with such an approach is that there is no natural way to determine which measure goes in the x or y column. Correlations are normally between different variables (e.g. height and weight). In a case of repeated measures, there is no natural order, so the intraclass correlation (ICC) is the more correct way to measure the similarity of within-group measurements. As ICC depends on a normal distribution, datasets must first be examined, and if necessary, transformed beforehand.
Robust methods have the advantage of permitting the use of untransformed data, which is especially useful when running scripts across hundreds of screening dataset features. The algorithm we use calculates a robust approximation of the ICC by combining resampling and non-parametric correlation.
Here is the algorithm, in a nutshell:
Group observations (e.g. cell count) by the grouping variable (e.g. target gene or antisense seed)
Randomly assign one value of each group to the x or y column (groups with one 1 observation are skipped)
for example, if the grouping variable is target gene and siRNAs targeting PLK1 had the values 23, 30, 37, 45, the program would randomly choose 1 of the values for the x column and another for the y column
Calcule Spearman’s rho (non-parametric measure of correlation)
Repeat steps 1-3 a set number of times (e.g. 300) and store the calculated rho’s
Calculate mean of the rho values from 4. This is the robust approximation of the ICC (rICC).
Values from 4 are also used to calculate confidence intervals.
The program that calculates this is available upon request.
Notes on calculating delta-delta G
Delta-delta G was calculated using the Vienna RNA package, as detailed here: https://www.biostars.org/p/58979/ (in answer by Brad Chapman).
The delta-delta G was calculated using 3 terminal bps. We found that that ddG of the terminal 3 bps had the strongest correlation with observed knockdown. Others (e.g. Schwarz et al., 2003 and Khvorova et al., 2003) have also used the terminal 4 bps.
Want to receive regular blog updates? Sign up for our siTOOLs Newsletter:
A recent article in The Scientist asks whether, in light of a paper by Lin et al. showing phenotypic discrepancies between RNAi and CRISPR, this is not ‘the last nail in the coffin for RNAi as a screening tool’?
The paper in question found that a gene (MELK) that had been shown by many RNAi-based studies to be critical for several cancer types shows no effect when knocked out via CRISPR. They also report that in relevant published genome-wide screens, MELK was not at the top of the hit lists.
Does this mean that the papers that used RNAi were unlucky and off-target effects were responsible for their observed phenotypes?
Gray et al. identified MELK as a gene of interest based on microarray experiments. They then designed RNAi experiments to test its role in proliferation. Assuming that this study and the subsequent ones followed good RNAi experimental design (using reagents with varying seed sequences, testing the correlation between gene knockdown and phenotypic strength, etc.), we can be fairly confident that MELK is involved in proliferation. It might not be the most essential player, which would explain why it is not at the top of screening hit lists. And screening lists have the draw-back of enriching foroff-target hits.
Another possibility is that Lin et al. have observed a known complicating feature of knock-out screens: genetic compensation. Although they undertake experiments to address this issue, it could be that compensation takes place too quickly for their experiments to rule it out. Furthermore, they could have addressed this issue by testing knock-down reagents themselves, and checking whether genes they hypothesise as responsible for the supposed off-target effect in the published RNAi work are in fact down-regulated. C911 reagents could also be used to test for off-target effects. This is extra work, but given that they are disputing the results in many published studies, this seems justified.
As regards the role of RNAi in screening, The Scientist concludes with the following (suggesting that their answer to the question of whether this is the final nail is also No):
In the meantime, one obvious solution to the problem of target identification and validation is to use both CRISPR and RNAi to validate a target before it moves into clinical research, rather than relying on a single method. “We have CRISPR and short hairpin reagents for every gene in the human genome,” said Bernards. “So when we see a phenotype with CRISPR, we validate with short hairpin, and the other way around. I think that would be ideal.”
Although we agree that validating CRISPR hits with RNAi reagents is important (especially if drugability is a concern), one has to be careful with RNAi reagents, like single siRNAs/shRNAs or low-complexity pools, that are susceptible to seed-based off-target effects. For validating CRISPR screening hits, siPOOLs provide the best protection against unwanted off-target effects, saving you time, money, and disappointment during the validation phase.
Want to receive regular blog updates? Sign up for our siTOOLs Newsletter:
In our last blog entry, we discussed a classic RNAi screening paper from 2005 that showed that the top 3 screening hits were were due to off-target effects.
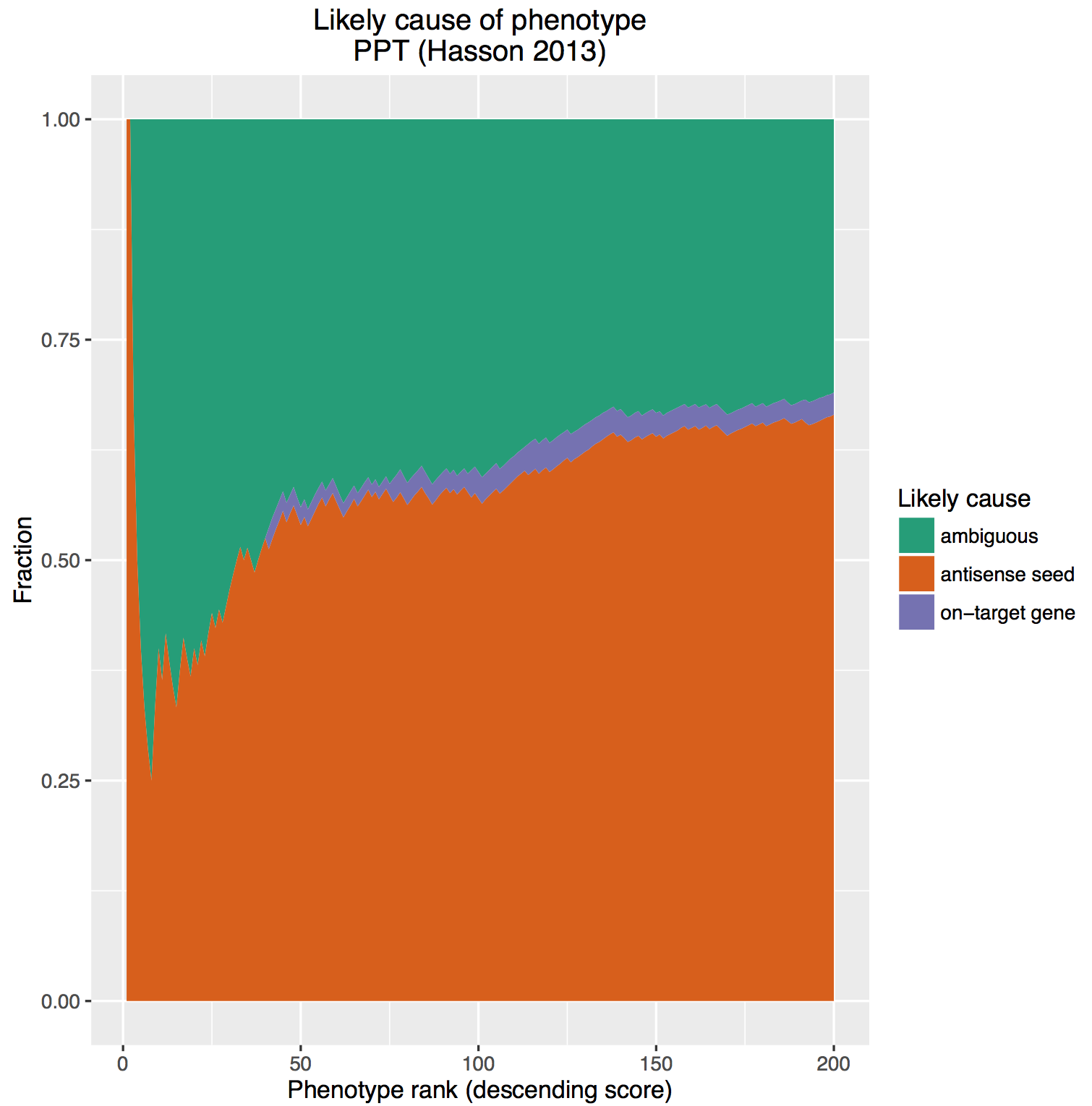
In this post, we analyse a more recent genome-wide RNAi screen by Hasson et al., looking in more detail at what proportion of top screening hits are due to on- vs. off-target effects.
Hasson et al. used the Silencer Select library, a second-generation siRNA library designed to optimise on-target knock down, and chemically modified to reduce off-target effects. Each gene is covered by 3 different siRNAs.
To begin the analysis, we ranked the screened siRNAs in descending order of % Parkin translocation, the study’s main readout.
We then performed a hypergeometric test on all genes covered by the ranked siRNAs. For example, if gene A has three siRNAs that rank 30, 44, and 60, we calculate a p-value for the likelihood of having siRNAs that rank that highly (more details provided at bottom of this post). It’s the underlying principle of the RSA algorithm, widely used in RNAi screening hit selection. If the 3 siRNAs for gene B have a ranking of 25, 1000, and 1500, the p-value will be higher (worse) than for gene A.
The same type of hypergeometric testing was done for the siRNA seeds in the ranked list. For example, if the seed ATCGAA was found in siRNAs having ranks of 11, 300, 4000, and 6000, we would calculate the p-value for those rankings. Seeds are over-represented in siRNAs at the top of the ranked list will have lower p-values.
After doing these hypergeometric tests, we had a gene p-value and a seed p-value for each row in the ranked list. We could then look at each row in the ranked list estimate whether the phenotypic is due to an on- or off-target effect by comparing the gene and seed p-values. [As a cutoff, we said that the effect is due to one of either gene or seed if the difference in p-value is at least two orders of magnitude. If the difference is less than this, the cause was considered ambiguous.]
After assigning the effect as gene/seed/ambiguous, we then calculated the cumulative percent of hits by effect at each position in the ranked list. Those fractions were then plotted as a stacked area chart (here, looking at the top 200 siRNAs from the screen):
The on-target effect is sandwiched between the massive ‘bun’ of off-target effects and ambiguous cause. We are reminded of these classic commercials from the 80s:
Want to receive regular blog updates? Sign up for our siTOOLs Newsletter:
Note on p-value calculations:
P-values were calculated using the cumulative hyper-geometric test (tests the probability of finding that many or more instances of members belonging to the particular group, in our case a particular gene or seed sequence). The p-value associated with a gene or seed is the best p-value for all the performed tests. For example, assume a gene had siRNAs with the following ranks: 5, 20, 1000. The first test calculates the p-value for finding 1 (of the 3) siRNAs when taking a sample of 5 siRNAs. The next test calculates the p-value for finding 2 (of 3) siRNAs when taking a sample of 20 siRNAs. And the last is the probability of getting 3 (of 3) siRNAs when taking a sample of 1000. If the best p-value came from the second test (2 of 3 siRNAs found in a sample size of 20), that is the p-value that the gene receives. This is also the approach used by the RSA (redundant siRNA activity) algorithm. One advantage of RSA is that it can compensate for variable knock down efficiency of the siRNAs covering a gene (e.g. if 1 of 3 gives little knockdown).
Like what you see? Mouse over icons to Follow / Share


 for Ambion vs. Dharmacon siRNAs grouped by same 7mer seed")