Tips for optimizing RNA affinity purification
RNA affinity purification (RAP) experiments enable the isolation and analysis of interacting molecules with an RNA of interest. Often performed to gain insight into RNA function, it is gaining popularity in the study of lncRNAs but can also be applied to coding RNAs.
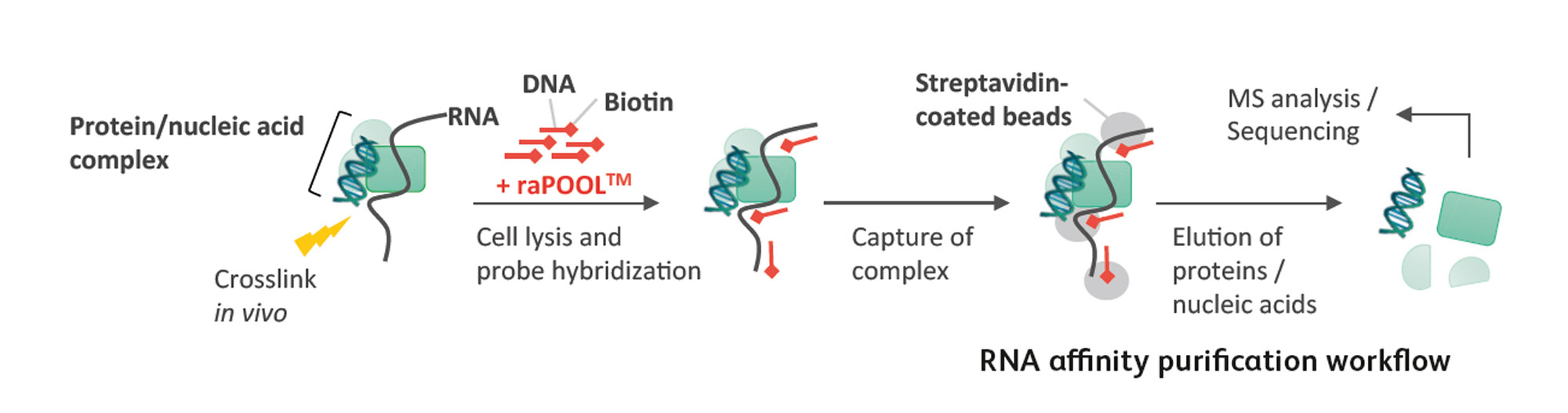
The general workflow involves preserving nucleic acid and protein interactions with a cross-linking reagent, followed by lysis and sonication to shear nucleic acids to sizes amenable for pulldown. Biotin-labelled DNA probes (which we offer, see raPOOLs) are added to the lysates and hybridize with the RNA of interest. This enables the isolation of the RNA and its associated molecules through the high affinity biotin-streptavidin interaction between biotinylated probes within the complex and streptavidin-coated magnetic beads. The complexes are then disrupted and individual components analysed by various methods: western blotting/mass spectrometry (for proteins), sequencing/northern and southern blots/PCR detection (for nucleic acids). A detailed protocol can be found here.
Despite the seemingly simple workflow, there are a number of parameters requiring optimization when applying the protocol to your RNA of interest. We break them down into the following sections:
1. Input material
If trying to detect protein partners of the target RNA, sufficient amounts of target RNA have to be isolated as unlike nucleic acids, proteins cannot be amplified. It is always advisable to determine the number of copies of the target RNA per cell to examine how much input material is required. As a guide, XIST is expressed at < 2000 copies/cell and required 50-250 million cells (depending on cell type) per pulldown condition to visualize proteins by immunoblotting/MS (Chu et al., 2015). For some lowly expressed lncRNAs, up to 1 billion cells may be required per pulldown! If your RNA is too lowly expressed and RAP is not feasible, other methods involving FISH and immunofluorescence imaging or protein arrays might be explored.
2. Cross-linking
Cross-linking reagents produce covalent bonds between nucleic acids and proteins in close proximity with each other, stabilizing these interactions for subsequent analysis. Formaldehyde (CH2O) and glutaraldehyde (C5H8O2) are most commonly used and cross-link direct and indirect associations between proteins-proteins, nucleic acids- nucleic acids and nucleic acids-proteins. Compared to formaldehyde, glutaraldehyde has two reactive groups, making it a stronger cross-linker. It also fixes long range interactions compared to formaldehyde, a zero-length cross-linker. Formaldehyde fixation can be reversed by heating at 65◦C for 6 h, while glutaraldehyde fixation is irreversible. Alternatively, UV irradiation produces irreversible cross-links only between nucleic acids and proteins.
There are a whole range of other cross-linking chemicals one could use in combination that can cross-link different reactive groups and at different ranges, but take note that the activity of cross-linking chemicals is highly determined by pH and buffer conditions, so be sure to follow given protocols closely and ensure reagents used are fresh. Note also that the longer cross-linking is carried out, the harder it is to sonicate nucleic acids down to smaller fragments.
3. Sonication and lysis
Sonication is an effective way to randomly shear nucleic acids to increase efficiency of their pulldown, hence improving detection sensitivity. Ideally, fragments should be sheared down to ~100-500 bases. If using PCR detection, be aware that your amplicon size should be small enough to lie within these sheared fragments.
Factors affecting sonication efficiency include volume of sample, sonication strength, frequency and duration and if using probe sonicators, probe position/depth. Follow the instrument manufacturer’s instructions closely and optimize duration of sonication by collecting samples at time-points and studying the extent of fragmentation. If using probe sonicators, ensure the probe is inserted into the lysate deep enough as frothing tends to occur during the sonication process. Always make sure to wipe the probe clean carefully each time. Due to the length of the protocol, take the usual precautions to avoid RNA degradation i.e. use RNase inhibitors, filter tips, and ensure temperatures are kept < 10°C. Lysates should be non-sticky and clear after sonication, which may take several hours. Pauses between sonications should be incorporated to avoid overheating of the sample.
If you are more focused on isolating chromatin, take note that lysis buffer conditions may vary to ensure efficient lysis of nuclei. In this case, swelling buffers may be incorporated and additional lysis steps might be required. Refer Chu et al., 2011.
4. Probe hybridization
The amount of biotinylated probes (raPOOL) should exceed the copy number of target RNA present in the lysate. With a recommended addition of 100 pmol of raPOOL per ml of lysate,
the number of copies of raPOOL =100 x 10^-12 * 6×10^23 (Avogadro’s constant) = 6 x 10^13 copies
and the number of copies of each probe (30 probes/raPOOL) = 6 x 10^13/30 = 2 x 10^12 copies
which is usually more than sufficient. However one can perform the hybridization with a range of probe concentrations to determine the optimal condition. Hybridization temperatures can also be varied with higher temperatures known to increase stringency of probe association.
5. Elution of components
As mentioned before, detection sensitivity is lowest for proteins, hence aliquot the beads in a proportion where most are used for protein analsis with a small proportion for nucleic acid detection. Benzonase is used to remove nucleic acids leaving proteins intact which can then be precipitated with TCA in the presence of deoxycholate. An acetone wash usually follows to remove the deoxycholate. Alternatively, biotin elution can be performed to compete out streptavidin binding sites and release complexes. It is recommended to separate the isolated proteins by PAGE to simplify the sample for MS analysis.
6. How much pulldown is enough?
The efficiency of RNA enrichment is determined by the difference in amounts of target RNA in the pulldown fraction as compared to the input fraction (i.e. lysate post-sonication and prior to probe hybridization). Be sure to account for the fraction of input used in the analysis. For a calculation guide, download this sheet. Fold enrichment is obtained by comparing against a negative/background condition. This may be performed using a negative control raPOOL which targets sequences not found in the cell. Non-target genes such as GAPDH/Cyclophilin A can also be analysed in the pulldown and input conditions to indicate pulldown is specific for the target RNA.
As a guide to how much pulldown is enough, Chu et al. retrieved ~60% of their target RNA and were able to analyse isolated proteins by western blotting and MS. One could also measure target RNA in lysates that have been subjected to pulldown, and a corresponding depletion of the target RNA should be observed.
We hope this helps you in your RAP optimization. For more questions and raPOOL requests, please feel free to email us at info@sitools.de
Want to receive regular blog updates? Sign up for our siTOOLs Newsletter: