Performing target validation well
Summary
This blogpost describes issues encountered in target validation and how to safeguard against poor reproducibility in RNAi experiments.
The importance of target validation
More than half of all clinical trials fail from a lack of drug efficacy. One of the major reasons for this is inadequate target validation.
Target validation involves verifying whether a target (protein/nucleic acid) merits the development of a drug (small molecule/biologic) for therapeutic application.
Failing to adequately validate a target can burden a pharma with roughly 800 million to 1.4 billion in drug development costs. Impact is not only monetary as large site closures often result as companies struggle to save costs and a reduced production effort deprives patients of new medicines.
Performing target validation well
Special attention should therefore be given to performing target validation techniques well.
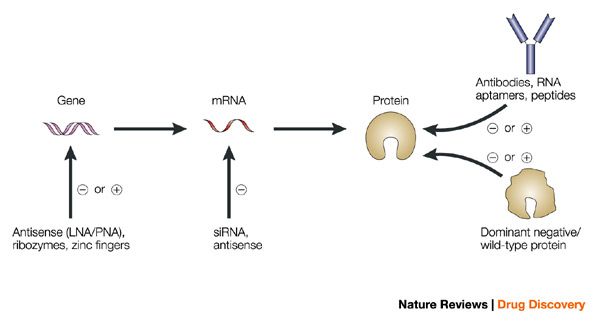
Many of these techniques involve inhibiting target expression to establish its relevance in a cellular or animal disease model. This can be performed with chemical probes, RNA interference (RNAi), genetic knock-outs, and even targeted protein degradation.
The reproducibility of these techniques however has been an issue of concern for drug developers. Less than half of all findings from peer-reviewed scientific publications was reported to be successfully reproduced.
Dismal rates of reproducibility from several pharma-led cancer-focused studies ranged from 11% (Amgen) to 25% (Bayer). A review by William Kaelin Jr sums up the common pitfalls of preclinical cancer target validation. One of his key points:
Cellular phenotypes caused by a chemical or genetic perturbant should be considered to be off-target until proved otherwise, especially when the phenotypes were detected in a down assay and therefore could reflect a nonspecific loss of cellular fitness. It is only by performing rescue experiments that one can formally address whether the effects of a perturbant are on-target.
The comment highlights the issue of reagent non-specificity as a notable contribution towards poor reproducibility.
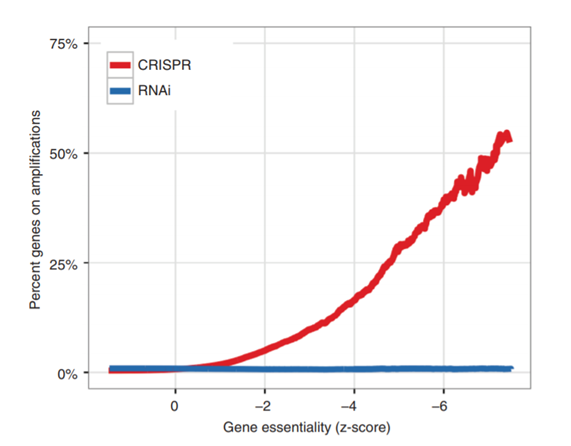
Certainly, for RNAi the wide-spread off-target effects of siRNAs has been observed in numerous publications. The mechanism being well-established to be based on microRNA-like seed-based recognition of non-target genes. The effect dominates over on-target effects in many large RNAi screens, illustrating the depth of the problem.
Reagent non-specificity is not restricted to RNAi. There have been multiple reports of non-specificity for gene editing technique, CRISPR, which can be read about in detail here, here and here. Recent publications continue to shed more light on its potential off-targets as we learn more about this relatively new technique.
Even chemical probes may have multiple targets. It is hence imperative that more than one target validation technique be used to avoid confirmation bias.
Target validation – a story from Pharma
Back in 2013, when siTOOLs was just starting out, a pharma approached us with a target validation problem.
They were obtaining different results with 3 different siRNAs in a cellular proliferation assay. Despite all 3 siRNAs potently downregulating the target gene, they produced different effects on cell viability.
Which siRNA tool to trust?
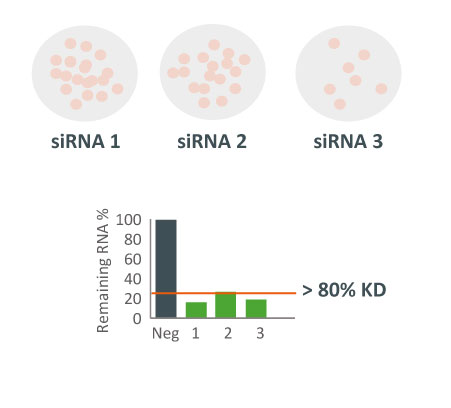
A whole-transcriptome expression analysis performed for the 3 siRNAs and a siPOOL designed against the same target revealed the reason for the large variability.
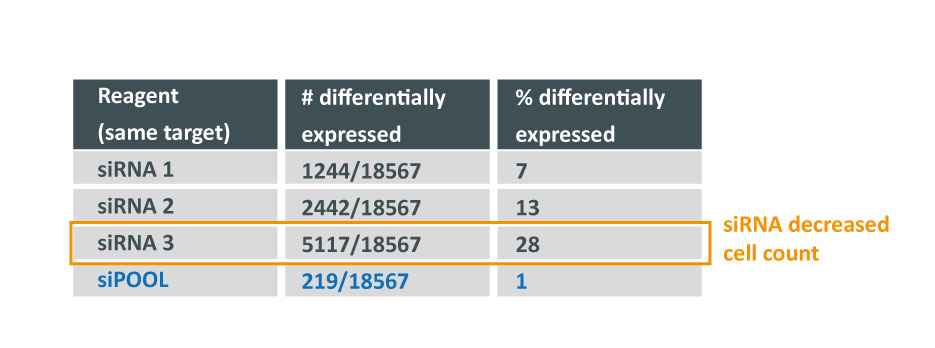
Despite all siRNA tools affecting the same target, the difference in extent of gene deregulation was astounding. With the greatest number of off-target effects, it was not surprising that siRNA 3 showed an impact on cell proliferation.
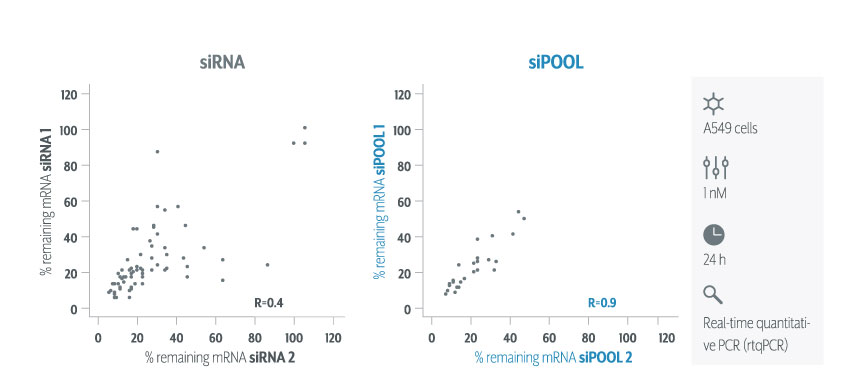
In contrast, siPOOLs had 5 to 25X less differentially expressed genes compared to the 3 commercial siRNAs against the same target. An expression analysis carried out for another gene target showed similar results i.e. siPOOLs having far less off-targets.
The target was dropped from development. A great example where failing early is a good thing, though it was not without costs from validating the multiple siRNAs.
The recommended target validation tool
Functioning like a pack of wolves, siPOOLs increase the chances of capturing large and difficult prey, while making full use of group diversity to compensate for individual weakness.
siPOOLs efficiently counter RNAi off-target effects by high complexity pooling of sequence-defined siRNAs. This enables individual siRNAs to be administered at much lower concentrations, below the threshold for stimulating significant off-target gene deregulation. Due to having multiple siRNAs against the same target gene, target gene knock-down is maintained and in fact becomes more efficient.
We still recommend using multiple target validation techniques. As a first evaluation however, siPOOLs are quick, easy and most of all, reliable.
Rescue experiments can also be performed with siPOOL-resistant rescue constructs.