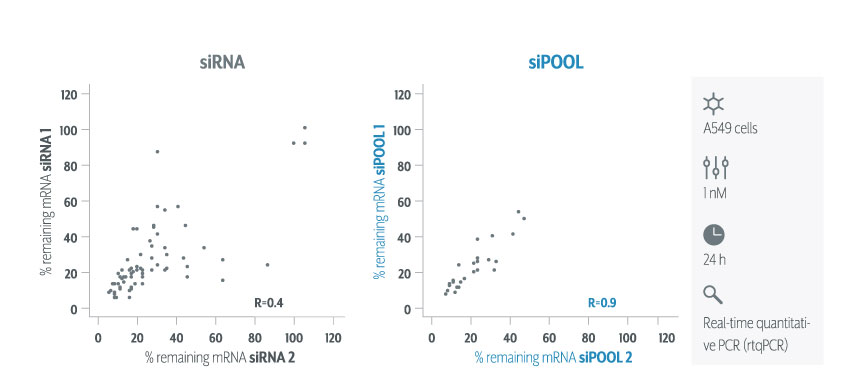
Short interfering RNA (siRNA) and short hairpin RNA (shRNA) are both used in RNAi-mediated gene silencing. In this blogpost, we explore the differences in applications of siRNA and shRNA and compare their capacity for off-targeting.
For a summary of their properties, please refer to Table 1 at the end of the post.
In what situations should we use siRNA or shRNA?
In terms of application, siRNAs are commonly applied for rapid and transient knockdown of gene expression.
It is performed in cell lines amenable to transfection by liposomes/electroporation and effects typically last from 3-7 days though retransfection can be performed to extend the effect.
The amount of siRNA introduced can be highly controlled and efficiency of gene knockdown is dependent on the levels of siRNA in the cell which is influenced by transfection efficiency and siRNA stability. Knockdown is also influenced by characteristics of the gene. A gene that is highly transcribed for example, may experience less siRNA-mediated downregulation compared to a gene where lesser copies of RNA are produced over time. In addition, a gene which expresses a protein with a very long half-life, may require extended periods of siRNA application to see a knockdown effect.
Due to the transient effect of siRNAs, shRNAs were developed to be used for prolonged knockdown of genes.
As they are introduced by viral vectors, cells that are more difficult to transfect are better targeted with shRNA. Furthermore, promoter-driven expression allows for inducible expression of the shRNA. Depending on the viral vector used – refer to Labome’s post that covers siRNA/shRNA delivery in greater detail – the shRNA may be integrated into the host genome, allowing it to be propagated into daughter cells. This maintains a consistent gene knockdown over several generations. However, knockdown efficiency can decline over time. This is mainly due to varying levels of uptake of the shRNA among cells, with a cell population having lower shRNA expression being over-represented with time.
What about RNAi screening?
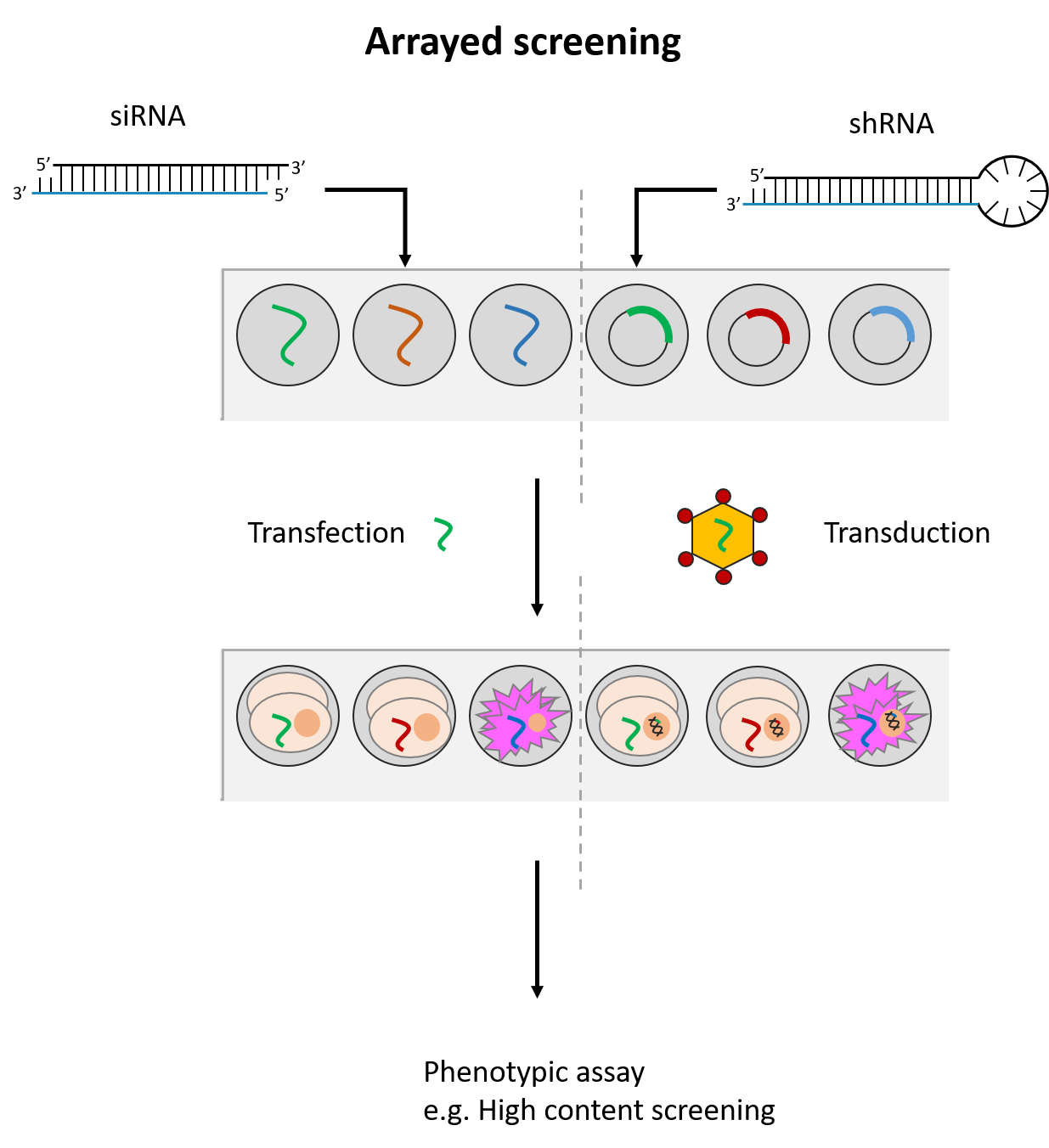
siRNAs and shRNAs are both used in RNAi screening to identify genes of interest in a studied phenotype. These are performed with siRNA/shRNA libraries that target a large variety of genes. There are two RNAi screening formats commonly used – arrayed and pooled.
siRNAs and shRNAs can both be used in an arrayed screening format. This means that the siRNA(s)/shRNA(s) against each gene is tested in distinct cell populations. Arrayed screens have the advantage of being compatible with various phenotypic readouts and do not suffer from possible reagent cross-talk or challenges associated with deconvoluting data. However, they are more energy and resource-intensive to perform. (See Fig. 2)
The pooled screening format in contrast, applies only with shRNAs. Here, all shRNAs (e.g. a whole-genome shRNA library) are introduced to a single cell population. As low titers of viral vectors are used, each cell in the population is expected to take up one shRNA vector.
With pooled screening, only readouts linked to cell number can be assessed. These include measurements for cell viability or altered expression of a cell surface marker assessed by fluorescence activated-cell sorting. shRNAs targeting genes which impact these readouts are expected to skew the cell population, such that only cells affected by the relevant shRNAs can be identified. This is either through negative selection, where lost cell populations are noted, or positive selection, where cells with certain shRNAs become over-represented.
The resulting cell population is then assessed by PCR, microarray hybridization or next generation sequencing to measure which shRNAs are highly or lowly-represented. The shRNAs are identified usually by means of a DNA barcode present in the vector sequence. Of note, pooled screens take up less resources to perform but require longer assay times to allow for significant changes in the overall cell population to occur.
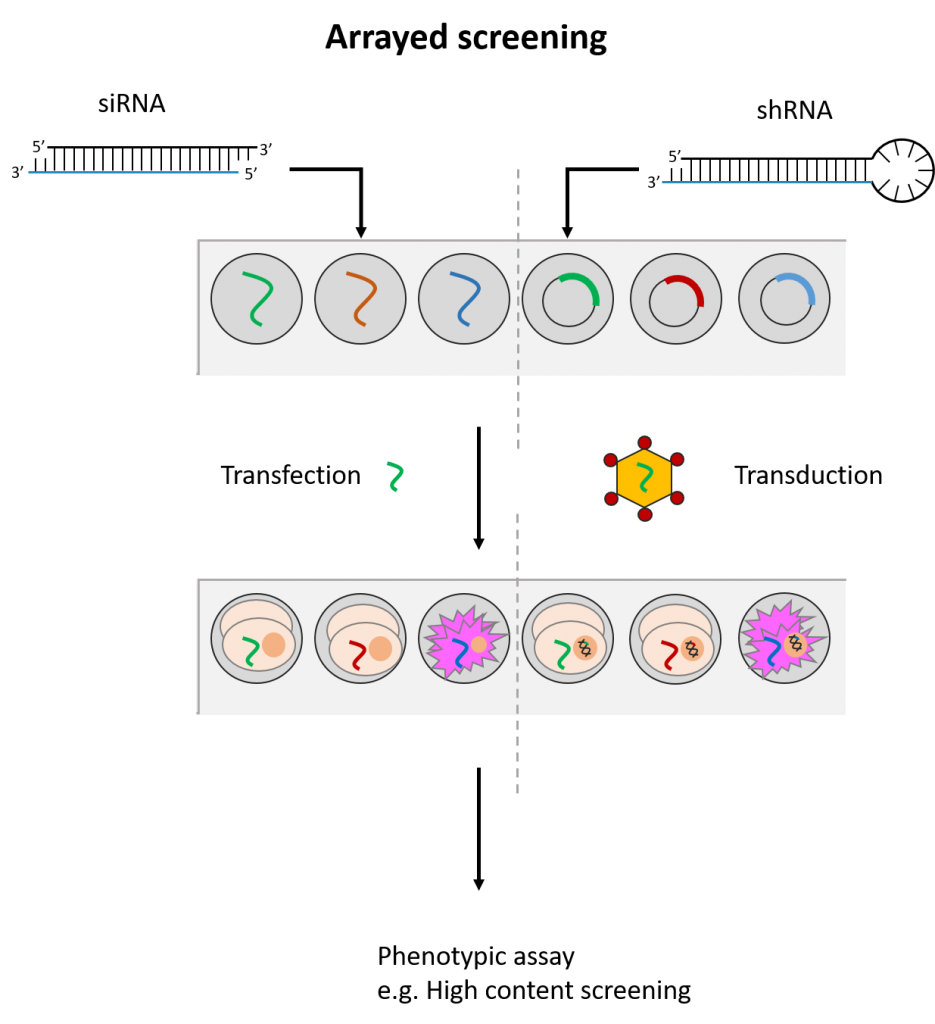
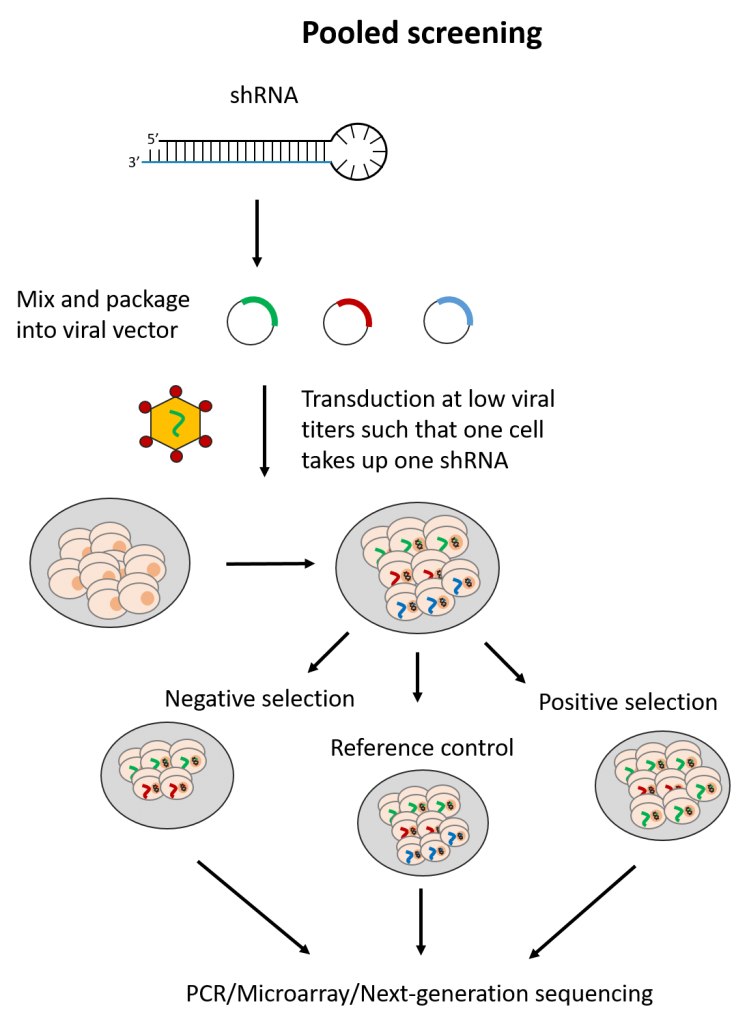
Fig. 2 Simplified workflow for arrayed and pooled RNAi screening formats
Off-target effects with shRNAs?
The use of siRNAs are known to produce several off-target effects but what about shRNAs? Given they are processed the same way as siRNAs, shRNAs are also subject to microRNA-like off-target effects. In addition, because they are expressed from DNA and rely on endogenous machinery to be processed into siRNA, several variations may be introduced not found with introducing siRNA directly. Some potential sources of off-target effects for shRNAs include:
1. Promoter-driven expression. shRNAs are typically controlled with a U6 promoter which drives high levels of transcription via RNA polymerase III. The high shRNA expression levels may saturate endogenous RNAi machinery, contributing to off-target effects. To counter this, shRNAs can be expressed in a context mimicking miRNAs, utilizing RNA polymerase II for transcription instead. This has been found by several groups to reduce the incidence of off-target effects (Grimm et al., 2006, Kampman et al., 2015)
2. Dicer-mediated hairpin processing. shRNAs undergo Dicer-mediated cleavage in the cytosol to remove its hairpin loop. Gu et al., 2012 reported that Dicer cleaves with sufficient heterogeneity to generate multiple sequences. This factor was reported to generate the higher noise levels unique to shRNA screens (Bhinder and Djaballah, 2013). As specificity of Dicer cleavage is influenced by neighbouring loop and bulge structures, care should be taken in shRNA design.
3. Multiple shRNA uptake. During viral transduction, the viral titer is minimized to increase the probability that cells take up a single shRNA vector. However, this does not guarantee that multiple shRNA uptake will not occur. In this event, a combinatorial gene knockdown ensues resulting in a mixed phenotype that may generate false hits.
4. Differences in genomic integration between shRNAs. Varying efficiencies in transfection and genome integration between shRNAs may skew results to over-represent certain shRNAs over others, especially in pooled screens. Furthermore, integration into the host genome may disrupt the function of certain genes, producing more off-targets.
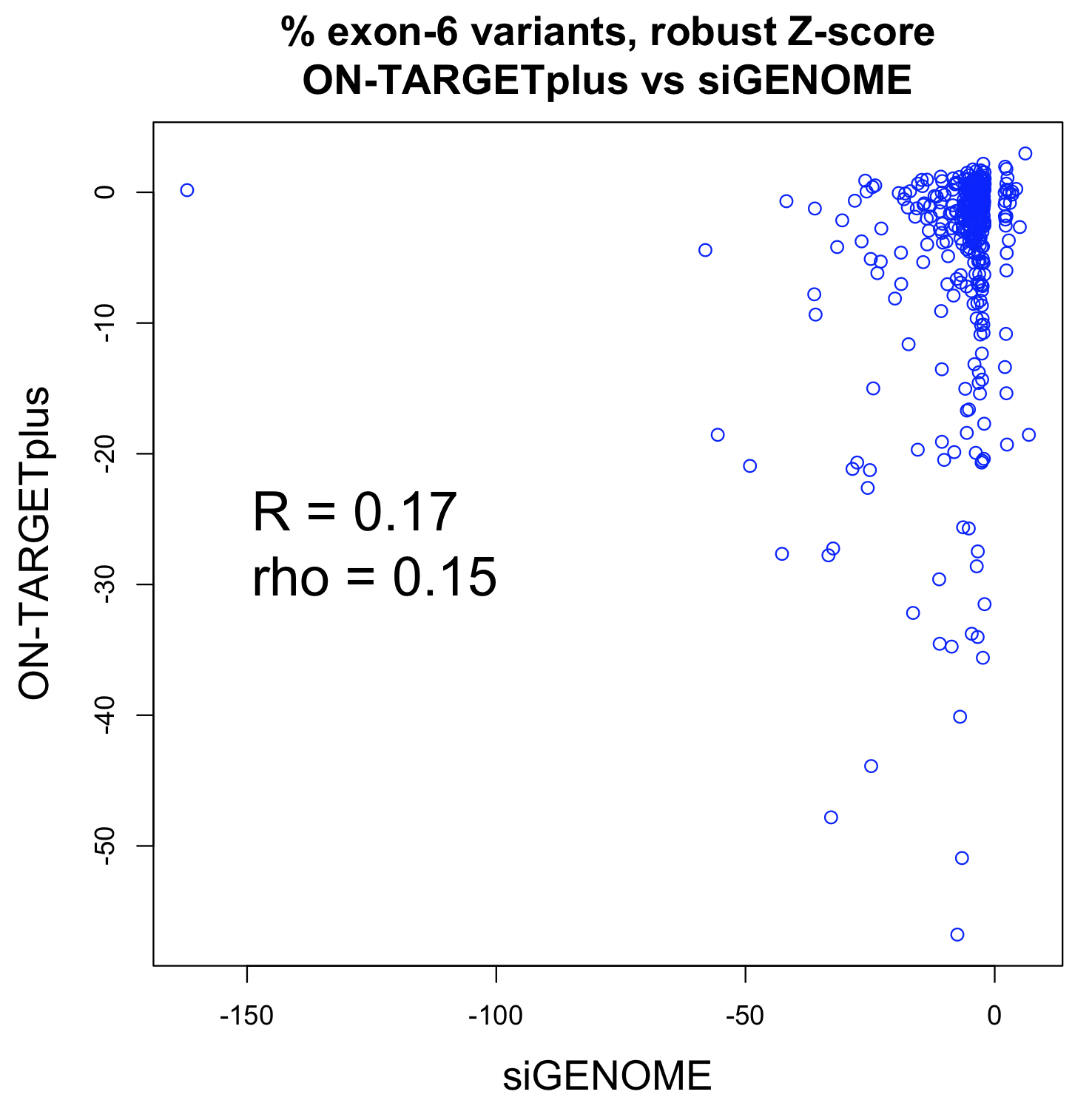
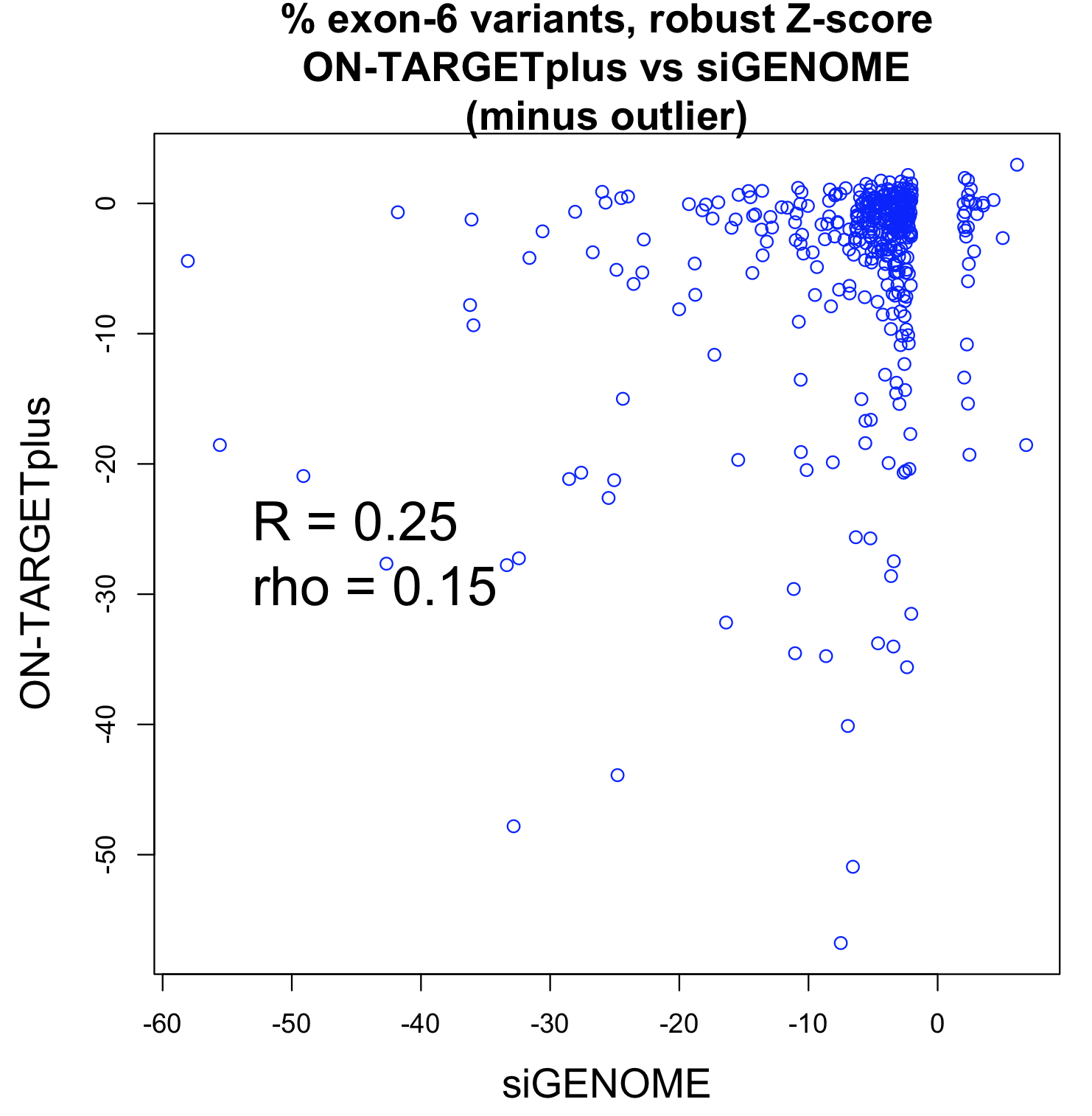
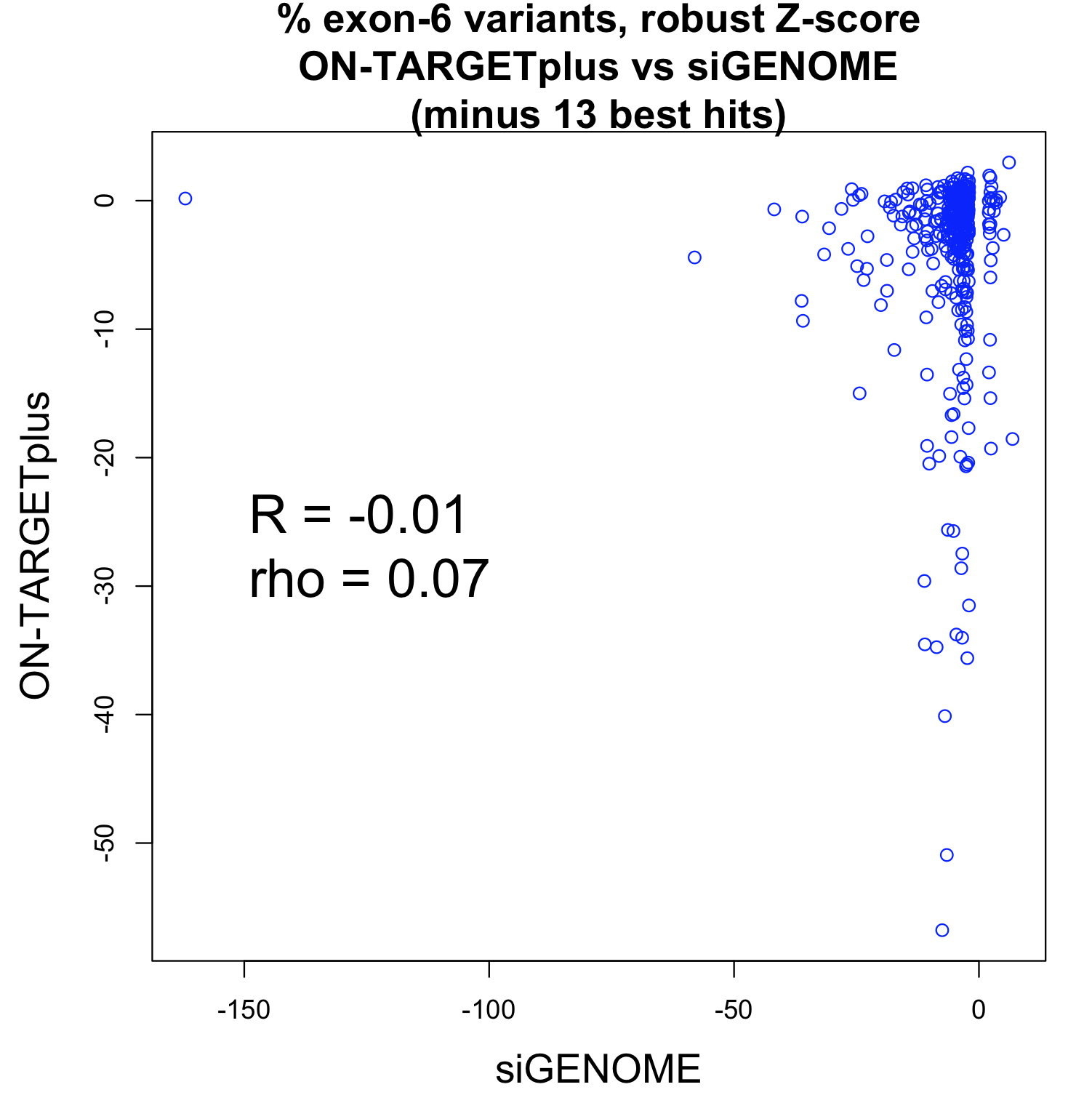
Studies comparing results from siRNA and shRNA screens have found extremely poor overlap, both between and within the reagent-specific screens. Bhinder and Djaballah’s (2013) analysis of results from 30 published RNAi screens (16 siRNA, 14 shRNA) searching for genes that impact cell viability saw no common genes identified across the board. Furthermore, different genes were identified depending on whether the screen used siRNA or shRNA. PLK1 for example, was a prominent hit for siRNA screens but was only marginally represented in shRNA screens. In contrast, KRAS was a top hit among shRNA screens.
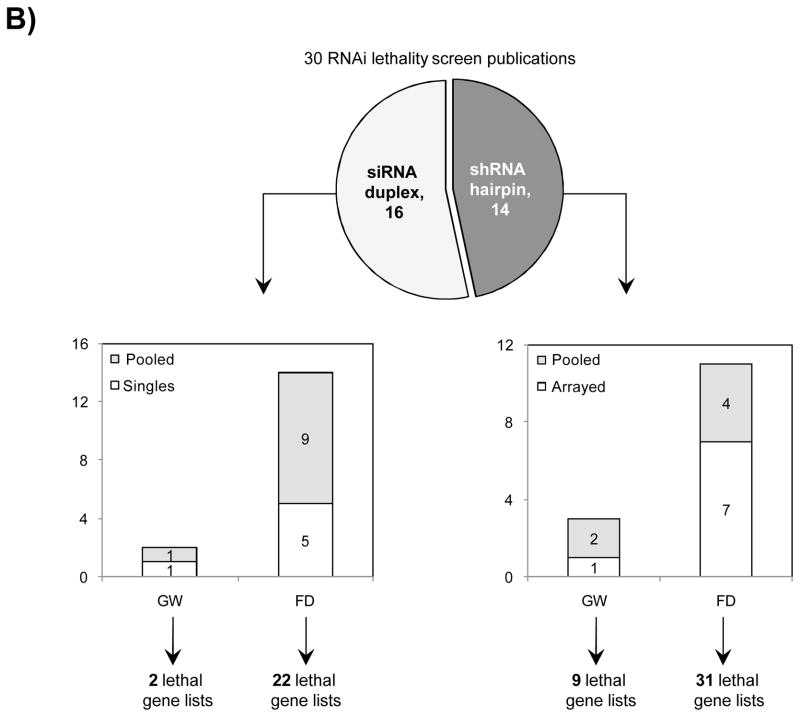
Fig. 3 Reagent format of RNAi screens analysed in Bhinder and Djaballah, 2013 Screens were performed either with genome-wide (GW) or focused (FD) siRNA/shRNA libraries. For siRNA screens, Pooled refers to pools of 3 siRNAs applied together compared to Singles where a single siRNA duplex was applied. For shRNA screens, Pooled refers to a pooled format screen (Fig. 2) where ~50, 000 shRNAs were applied to a single cell population. Arrayed refers to arrayed format screen where shRNAs were applied individually (Fig. 2).
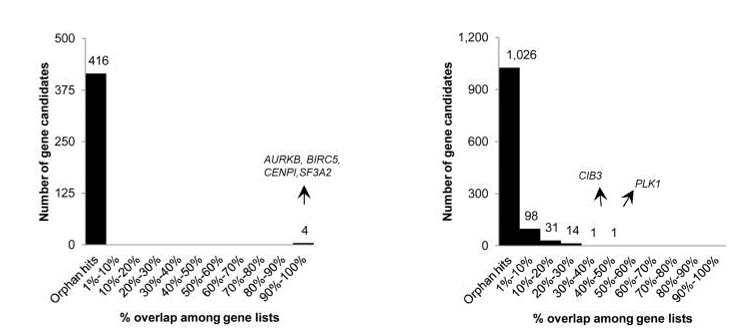
Fig. 4 Overlap of hits among genome-wide (left) and focused (right) siRNA screens (Bhinder and Djaballah, 2013) Only 4 common hits detected across the 2 lethal gene lists from genome-wide siRNA screens. In focused siRNA screens, a greater overlap was detected but still limited across the 22 lethal gene lists. PLK1 detected in 9 out of 22 gene lists.
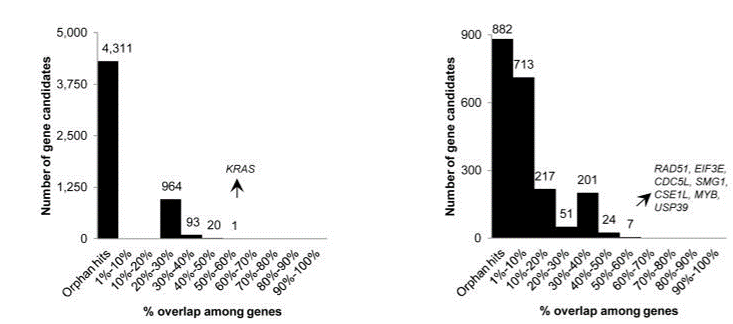
Fig. 5 Overlap of hits among genome-wide (left) and focused (right) shRNA screens (Bhinder and Djaballah, 2013) KRAS was a top hit in shRNA GW screens, appearing in 5 out of 9 lists. In focused shRNA screens, KRAS was present in 15 out of 31 lists.
Worryingly, an enrichment of gene candidates exclusive to pooled shRNA screens was observed as opposed to arrayed shRNA or siRNA screens. Most of the overlap seen in gene lists (80% global overlaps, 60% after stringent filtering) were specific to pooled shRNA screens. Exclusion of data from pooled shRNA screens would have reduced overlap to a mere 27%. This indicates gene targets obtained from shRNA pooled screens is specific to the technique as opposed to specific gene downregulation.
Furthermore, a greater number of hits were obtained from shRNA screens – 6664 candidates from 40 shRNA gene lists – as opposed to 1525 candidates from 24 siRNA gene lists. This indicates a generally noisier dataset associated with shRNA screens.
Bhinder and Djaballah later performed a head-to-head comparison of an arrayed siRNA and shRNA screen and reported similarly dismal results. Despite using a gain-of-function assay, which tends to yield clearer results, only a 29 hit overlap was seen between siRNA and shRNA libraries which shared 15,068 common genes. Based on a known set of positive controls, siRNAs identified 8 known regulators as opposed to shRNA which only identified 3. Furthermore, predicted siRNA sequences obtained after Dicer-processing of shRNA which corresponded to exactly the same siRNA sequence from the siRNA library yielded different phenotypes. The authors highlight that differential intracellular processing of the shRNA contributes significantly to the discrepancies observed.
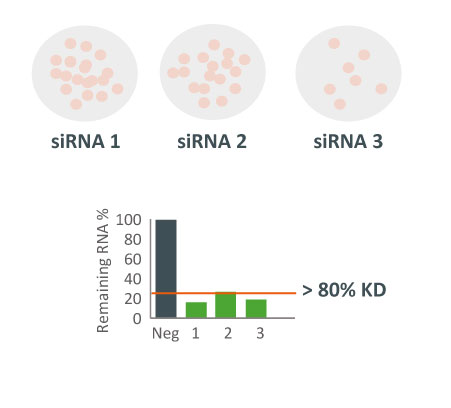
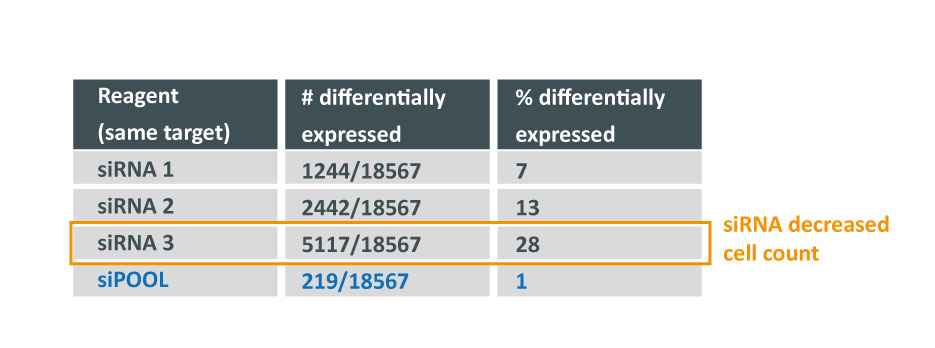
It is evident that shRNAs are at risk to greater number of off-target effects than siRNAs. Much care should be taken towards the interpretation of pooled shRNA screens in particular. Secondary validation of gene hits plays an increasingly important role. It is recommended to validate gene hits with siPOOLs (high-complexity, defined siRNA pools) which have a lower off-target profile than single siRNAs or low complexity siRNA pools of 3-4. siPOOL-resistant rescue constructs enable further affirmation that the loss-of-function phenotype is attributed to the target gene. Alternative tools such as compounds, antibodies or gene knockout technologies are also highly recommended.
Want to receive regular blog updates? Sign up for our siTOOLs Newsletter:
Table. 1 Comparison of properties between siRNA and shRNA
|
siRNA |
shRNA |
| Structure |
 20-25 nucleotide long double-stranded RNA (dsRNA) with 2 nucleotide overhangs at the 3’ end 20-25 nucleotide long double-stranded RNA (dsRNA) with 2 nucleotide overhangs at the 3’ end |

~57-58 nucleotide long RNA sequence with a dsRNA region linked by non-pairing nucleotides to form a stem-loop structure |
| Delivery |
RNA itself with liposome/electroporation-mediated delivery into cells |
Usually delivered to cells via viral vectors. DNA may be incorporated into host genome depending on viral vector used. |
| Processing |
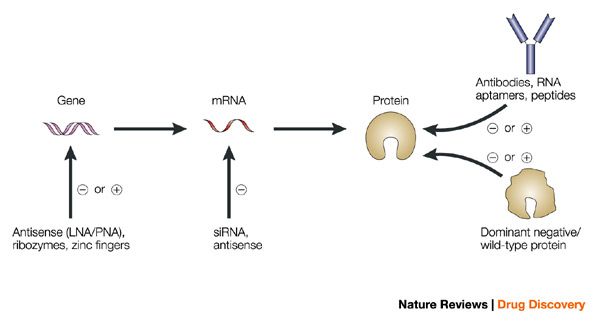
In the cytosol, guide or antisense strand* (shown in blue in Fig. 1) is incorporated into RNA induced silencing complex (RISC). RISC is guided towards RNA transcripts with the complementary sequence to mediate cleavage and subsequent degradation of the transcript.
*Note that the sense strand may also load into RISC and mediate off-targeting but incidence of this is reduced by designing siRNA with appropriate thermodynamic properties (refer to previous blogpost on siRNA design) |
In the nucleus, shRNA is transcribed from DNA by either RNA polymerase I or III, depending on the promoter.
Drosha, a member of the ribonuclease III family, processes the RNA transcript of its long flanking single-stranded RNA sequences and the resultant shRNA is exported out of the nucleus by Exportin-5.
In the cytosol, the enzyme Dicer cuts off the hairpin loop of the shRNA and releases the functional active siRNA which follows the same downstream processing as siRNAs. |
| Length of expression |
Varies from 3-7 days. Affected by degradation of siRNA within cell and dilution of effect upon cell division. Expression can be reinstated by re-transfecting the siRNA. |
If the DNA is stably integrated in the host genome, knock-down is theoretically permanent. |
| Control of knockdown |
Easily controlled by varying amount of siRNA introduced. |
Magnitude of knockdown harder to control as determined by promoter-driven efficiency and shRNA vector uptake. Expression however can be made inducible with Tet-on/off systems. |