A Brief Interview with Dr. Blaine Fritz
Chronic diseases like lower extremity wounds, which prevail in diabetic patients, are expensive to treat and cause reoccurring trauma to patients. Diabetic-related foot ulcers (DFUs) are also highly prone to infections, which increases the risk of hospital admission and amputation. Therefore, properly classifying infection severity in lower extremity ulcers is essential to guide appropriate treatment.
Presently, clinicians and researchers use subjective classification systems to stratify lower extremity ulcer infections for treatment and research. These classifications are based on guidelines that combine clinical data and wound observations. To better understand the relationship between infection severity and clinical classifications, a study by Fritz et al. (2022) focused on obtaining host and bacterial RNA sequences from infected human tissues using dual, host-pathogen RNA sequencing. The objective of this approach was to see whether these clinical classifications reflect the ulcer’s transcriptome. The results of the experiment (*SPOILER ALERT*) suggest that indeed stratification of infection status based on a transcriptomic fingerprint may be an objective method to categorize infection severity.

For more details results, you can read the full article here.
Now more on the scientist behind the research, Dr. Blaine Fritz (pictured on the right) is a post-doc working at the University of Copenhagen in the Department of Immunology and Microbiology. He is part of the Costeron Biofilm Center and has a background in studying host-pathogen interactions. We asked him a few questions to get to know more about him, his research and his motivation 😊.
1. What is it about bacteria that interests you the most?
The most interesting thing about bacteria, for me, is the huge role that they play in our everyday lives, which we are just beginning to discover. The advent of computational and molecular tools for understanding bacterial communities has allowed us a much deeper look into these microbial communities and their function.
- When did you start studying ulcer infections treatment and research?
I started studying bacterial infections in ulcers and various other diseases during my master and PhD work at the University of Copenhagen in 2014. Prior to that, I also worked with bacterial biofilms, but in an industrial rather than a clinical setting.
- How does RNA sequencing and thus gene expression in ulcer tissue and infecting bacteria help provide valuable information?
Our application of dual RNA sequencing allows an insight into physiology of both human cells and infecting bacteria as they are directly in an infection. This gives a picture of how the immune system responds to bacterial infection and vice versa.
- What is the direct impact your latest research findings will have on patients with chronic lower extremity wounds?
We hope that these results will help researchers and physicians understand the major factors of host response to bacterial infections during chronic ulcers. Though this is basic research, the methods and data are publically available for others to work further on this and eventually develop methods or targets for mitigating these chronic bacterial infections.
- What tools are key in your methodology?
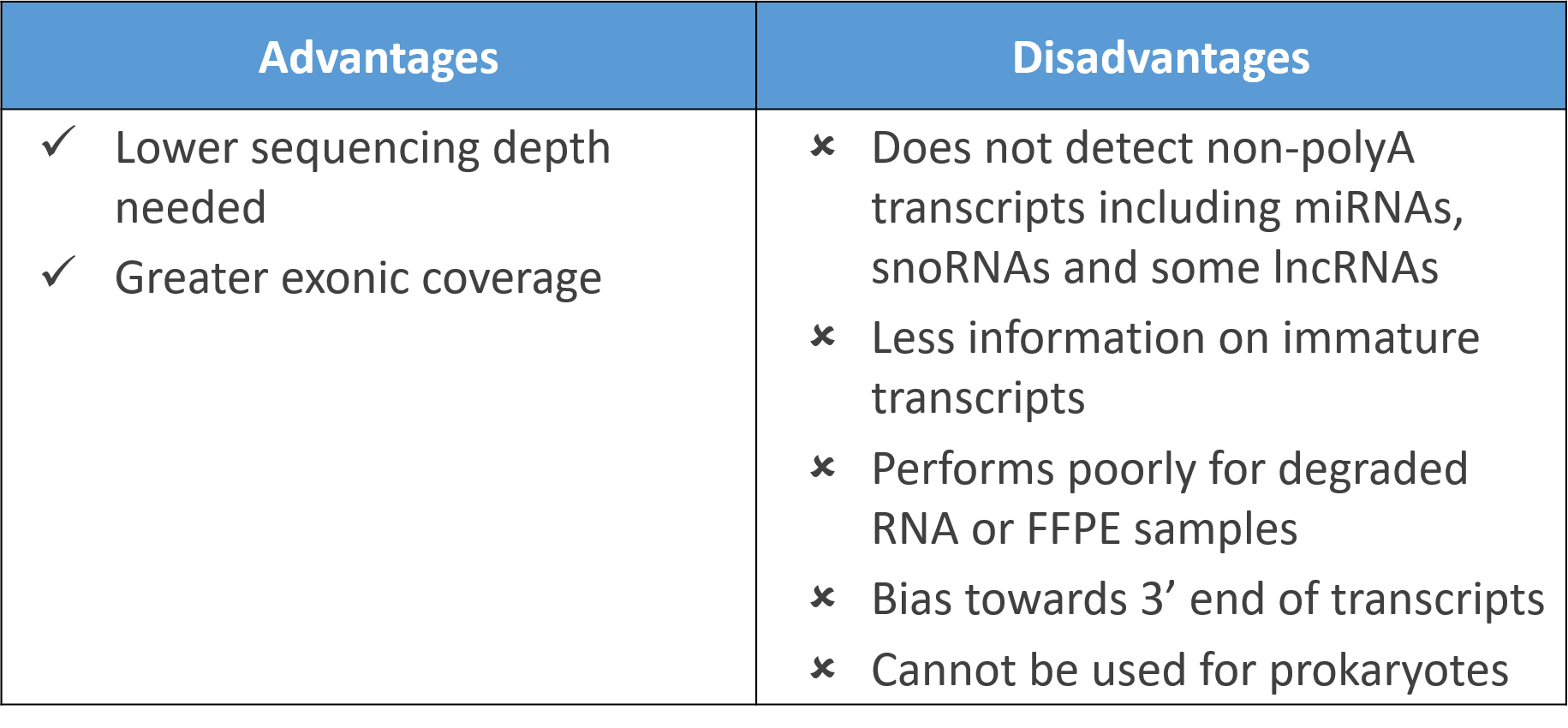
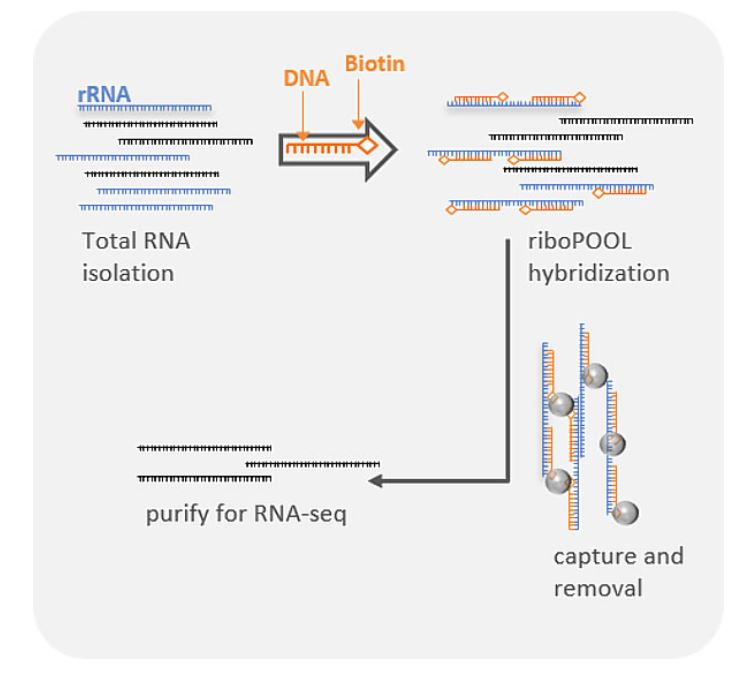
Several things – including the preparation of the sample for RNA-sequencing and the bioinformatics processing of the data are essential for our methodology. For example, a high sequencing depth in combination with performing ribosomal RNA depletion (in this case we used a mix of 10:1 Human:Pan-Prokaryote riboPOOLs) rather than poly-A enrichment is a key step to obtaining also bacterial RNA sequences from infected human tissues.
- What fascinates you the most about your job?
What fascinates me most is the incorporation of advanced technologies for sequencing data in combination with our increased access to high-performance computing in order to solve complex biological problems and ultimately improve patient livelihood. I think that the application of these technologies and others are just beginning to be explored.
- If you would have not been a scientist, what other profession would you have chosen?
I think, had I not become a scientist, I would have become an engineer. I am also interested in music, so I also thought about becoming a sound engineer or a professional musician.
- What is the best career advice you have gotten?
I think one of the best pieces of advice to remember is that it is YOU who is the asset. Always remember that you possess specialized skills. So, remember that it is not what you can do for your employer, but rather what your employer can do for you in order to make your working life enjoyable. Never get stuck in a career which doesn’t make you happy!
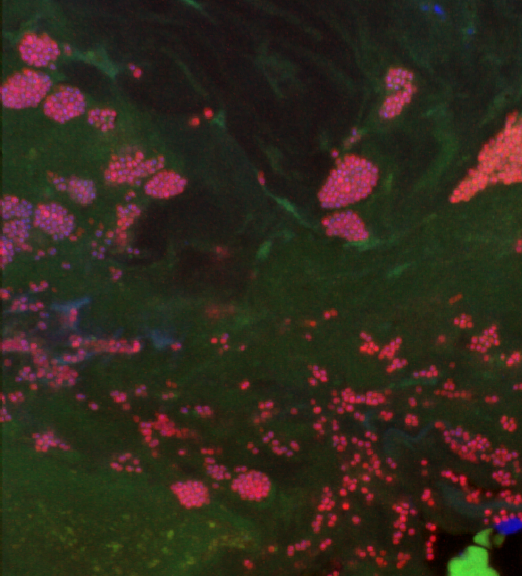
Image: Biofilm aggregates in an infection (provided by Dr. Fritz).





{kind=link}