
Understanding Gene Networks with Combinatorial Gene Knockdown
Genes hardly ever work alone, functioning instead in complex gene networks. Increasing advances in genomics and proteomics and corresponding developments in computational analysis, has really put this into perspective. A recent large scale RNAi study by Novartis found this hairball of a gene network in cancer cells:
A gene “hairball”
As such, the standard approach of disrupting the expression of a single gene to study loss-of-function phenotypes may not accurately reveal its genetic function. The highly redundant nature of signalling pathways often allows cells to respond robustly to single-strike manipulations. A combinatorial gene disruption approach, where one disrupts several genes in a single setting, is therefore more effective at elucidating signalling networks.

Types of redundancy
Redundancy can occur across signalling pathways or within a signalling pathway. Paralogous genes arising from gene duplication (B’) may also contribute to redundancy. In the figure above, gene B is disrupted but phenotype remains unaffected if other genes (A or B’) can perform similar functions. Within a single pathway, genetic interactions (A → C) may exist that make B redundant.
In addition to countering redundancy, combinatorial gene disruptions also uncover interesting epistatic or synergistic interactions. An epistatic or synergistic interaction occurs when the effect of disrupting two genes differs from the additive effect expected from disrupting the genes individually. This reveals the nature of genetic interactions and identifies interesting functional networks that play relevant roles in complex diseases. In cancer for example, a combinatorial approach is useful for identifying genes that confer drug resistance and to explore multi-treatment approaches that achieve synthetic lethality of cancer cells.
Combinatorial gene disruption has been successfully applied to yeast where multiple knockouts are easy to generate (Fiedler et al., 2009). However, multiple knockouts are harder to perform in higher organisms and may also not represent the full picture. RNAi, due to its ease of application, has been used for combinatorial disruptions in Drosphila (Nir et al., 2010, Horn et al., 2011), C. Elegans (Tischler et al., 2006) and human cells (Laufer et al., 2013). Its dose-dependency and transient effect also mimics the use of drugs and allows researchers to determine the quantitative nature of functional interactions.
Studies have found that predictions of genetic interactions made based on double gene knockdowns showed greater sensitivity than predictions based on single gene knockdowns. Nir et al analysed cell morphological changes under RNAi knockdown of RhoGAPs in a RhoGTPase overexpression condition. Using single/double knockdowns to validate 5 biologically validated interactions and 3 non-interactions, they found the double knockdowns were far more sensitive in detecting genetic interactions.
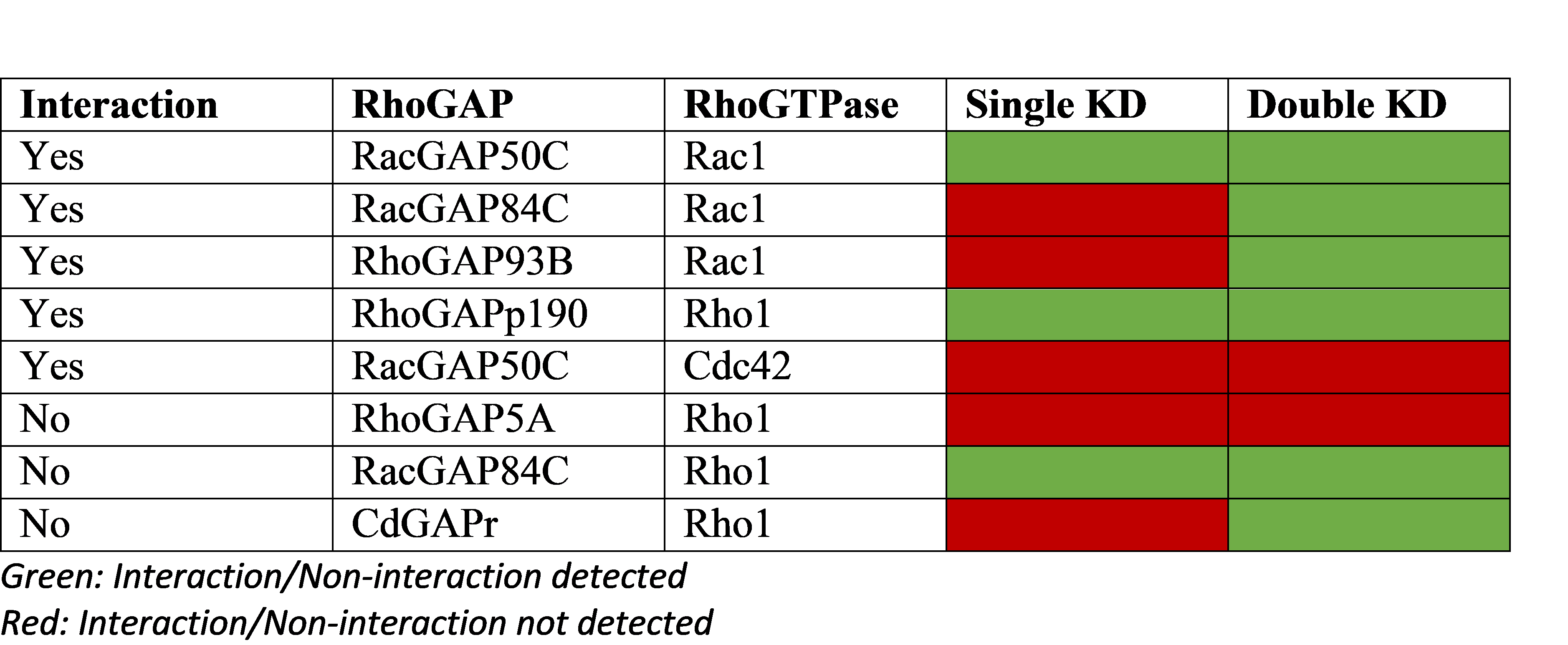 Double gene knockdowns (KD) improve sensitivity of genetic interaction detection
Double gene knockdowns (KD) improve sensitivity of genetic interaction detection
Relying on the fact that GAPs deactivate GTPases, a screen using single/double RNAi KDs was performed on RhoGAPs in Drosophila cells. The table above shows validated biological interactions (5 interactions, 3 non-interactions) and prediction success from the single/double KD experiments.
Larger scale studies looking at looking at 50 000 to 70 000 pairwise perturbations of signalling factors in both Drosophila (Horn et al., 2011) and human cells (Laufer et al., 2013) saw similar results. A higher sensitivity was afforded by the double knockdowns and phenotypes obtained from single knockdowns often differed from double knockdowns.
Some challenges highlighted from these large combinatorial RNAi studies:
- Inconsistent phenotypes from single siRNAs that target the same gene either due to off-target effects or poor knockdown (KD) efficiencies. This is a known problem with siRNAs that siPOOLs were developed to counter. In Laufer et al (2013), an additional quality control step had to be taken to remove inconsistent siRNAs and choose siRNAs that provided good KD.
- Need for large sample sizes. When Laufer et al. reduced the number of cells analysed from 7100 to 1775, the number of genetic interactions detected decreased from 5262 to 1022 indicating reduced sensitivity. This is naturally assay dependent as well with larger, robust phenotypes requiring smaller sample sizes. A multiparametric (measuring multiple parameters of cell behaviour/morphology) approach is often encouraged to increase data robustness.
- Differences between model organisms. Knockdown efficiencies in human cells were lower compared to Drosophila cells and off-target effects more widespread. This is a factor for consideration as additional computational analysis and reagent pre-evaluation may be necessary.
- Greater resources required performing double/triple knockdowns compared to single gene knockdowns. Furthermore, measuring these phenotypes in multiple cell lines are often recommended to affirm phenotypes. Therefore, a focussed approach looking at interesting subsets of genes is recommended.
- Risk of toxicity increases with increasing concentrations of siRNA used.
The use of siPOOLs counters some of the challenges faced in combinatorial RNAi knockdowns. Due to the low effective working concentration, multiple siPOOLs can be used together with reduced risk of toxicity. The lowered off-target profile and high reproducibility and robustness of on-target knockdown demonstrated with siPOOLs also add to greater data reliability and eliminates the need for siRNA pre-evaluation.
Dr. Derek Welsbie et al. from the University of California, San Diego, recently published in Neuron the use of siPOOLs in a combinatorial knockdown approach. A synergistic relationship between Leucine Zipper Kinase (LZK) and Dual Leucine Zipper Kinase (DLK) was identified to promote survival in an axon degeneration model with primary mouse retinal ganglion cells (RGCs).
A large high-throughput functional genomic screen where cells were first subject to DLK knockdown was performed to sensitize them to other kinase siRNAs that promote RGC survival. In this way LZK was identified and the synergistic relationship was verified with siPOOLs:
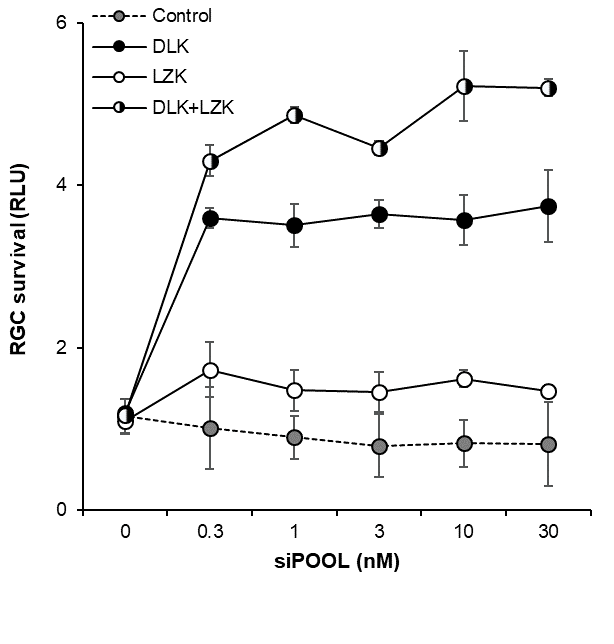
LZK and DLK synergize to promote retinal ganglion cell survival
Knockdown of LZK alone produced no visible effect but siPOOL-mediated knockdown of both LZK and DLK produced a synergistic effect on cell survival.
An additional screen was performed where LZK siPOOL was used to sensitize RGCs to protective effects afforded by DLK and potential novel DLK pathway members. Screening performed with 16 698 low complexity pools of 4 siRNAs each, identified 6 novel hits. Though these failed to be verified following siRNA deconvolution (learn why here and here), Haystack analysis to account for seed-based off-targets verified certain hits and additionally identified new hits such as Sox11.
siPOOL-mediated combinatorial knockdown of four identified genes – Sox11, Mef2a, Jun and Atf2 – highly promoted RGC survival under colchicine-induced injury. The survival-promoting synergistic effects of all four transcription factors was comparable to that of the DLK/LZK interaction.
Notably, these effects were verified with CRISPR sgRNA knockouts.
Combinatorial gene disruption allows us to learn more about gene networks and the nature of genetic interactions. Complementing gene knockout approaches, RNAi is an easy method of performing combinatorial gene disruptions in the transient setting.
siPOOLs afford the added advantage of increased efficiency and reliability, removing the need for siRNA pre-evaluation and increasing ease of data analysis.
For more relevant research/siTOOL updates, sign up for our siTOOLs Newsletter: