Ribo-depletion in RNA-Seq – Which ribosomal RNA depletion method works best?
Summary: This blogpost is focussed on ribosomal RNA (rRNA) depletion methods frequently applied to improve and economize RNA-Seq experiments.
The Rise of RNA-Seq
RNA-Seq Overtakes Microarrays
The use of Next-Generation RNA Sequencing (RNA-Seq) has recently overtaken that of DNA-based microarrays to detect and quantify changes in gene expression.
Why? RNA-Seq can detect novel coding and non-coding genes, splice isoforms, single nucleotide variants and gene fusions. Its broader dynamic range also enables sensitive detection of low abundance transcripts.

Also, technological advancements in single cell isolation, ribosome profiling and pulse-labelling techniques can now be multiplexed with RNA-Seq to widen the scope of scientific interrogation. Now, one can study the transcriptome, translotome and epitranscriptome with added spatial and temporal resolution. Studies of RNA structure and RNA-protein/nucleic acid interactions with the use of nuclease digestion and biochemical pulldown approaches have also increasingly employed RNA-Seq. This excellent review describes all these latest advances.
A Major Challenge in RNA-Seq
A major limitation encountered in RNA-Seq however is the massive abundance of ribosomal RNA (rRNA) that can occupy up to 90% of RNA-Seq reads. This necessitates additional steps for ribo-depletion or rRNA depletion to economize an RNA-Seq experiment.
Ribo-Depletion Methods
1) Poly-A selection
The most common method of rRNA depletion is poly-A selection, which relies on the use of oligo (dT) primers attached to a solid support (e.g. magnetic beads) to isolate protein-coding polyadenylated RNA transcripts. The main disadvantage though is one misses out on non-polyadenylated transcripts which include microRNAs, small nucleolar RNAs (snoRNAs), some long non-coding RNAs (lncRNA), and even some protein-coding RNAs (histones) which lack polyA tails. As a result, one fails to capture biologically relevant insights on these RNAs which make up a substantial proportion of the transcriptome.
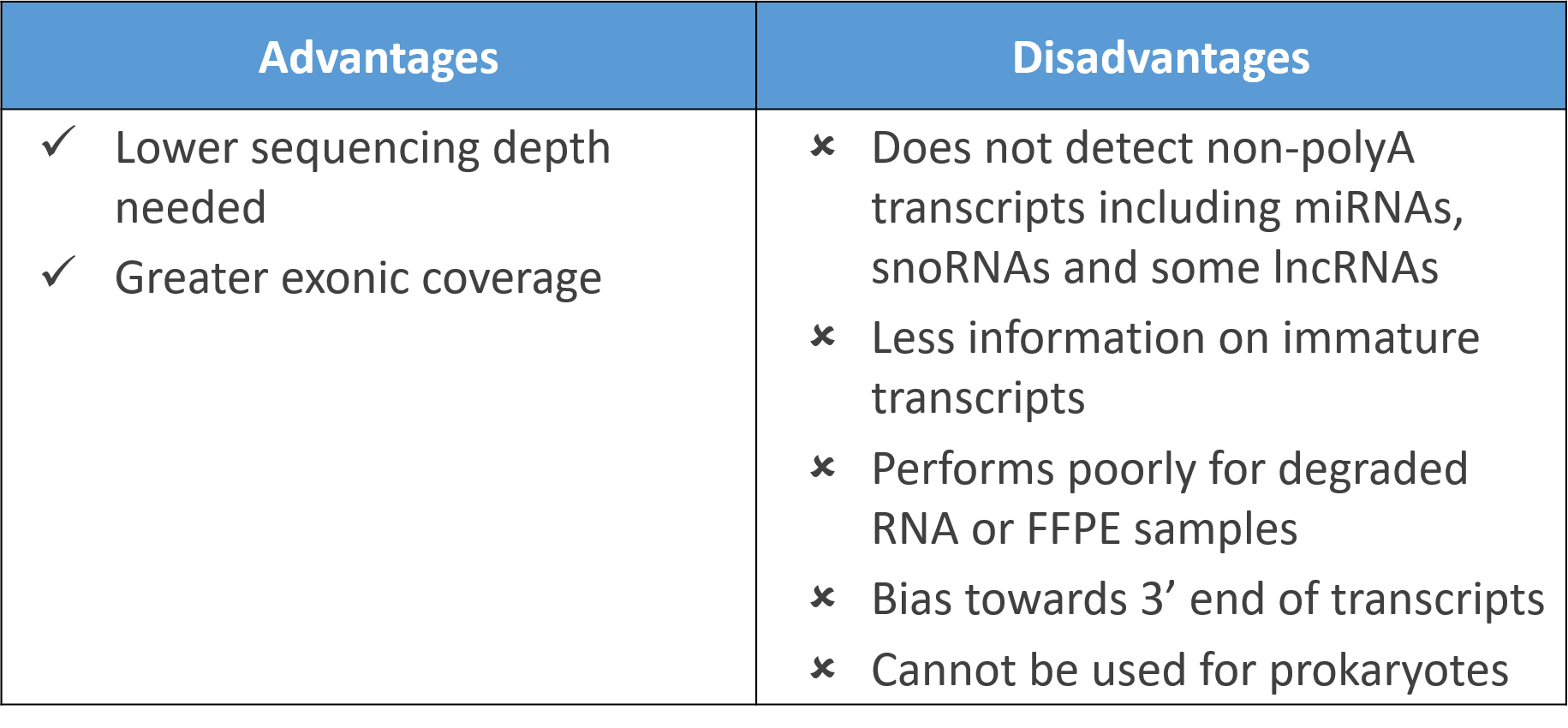

Curiously, polyadenylated transcripts are more abundant in eukaryotes as opposed to prokaryotes with both groups using polyadenylation in entirely different ways! Hence, polyA selection cannot be applied for sequencing of bacteria and archaebacteria, excluding its use in metagenomic RNA-Seq.
Poly-A selection also relies on transcripts being largely intact and tend to over-represent 3′ regions of transcripts. Studies comparing physical rRNA depletion methods and polyA selection show polyA selection did not work well for degraded RNA samples. A lower number of reads were also obtained with formalin-fixed paraffin-embedded (FFPE) tissues though analysis of fresh frozen tissues was not compromised.
Despite this, polyA selection still provides greater exonic coverage than physical rRNA depletion which tend to produce more intronic reads. Further, a lower sequencing depth is typically needed for polyA selection, making it a respectable choice if one is focused only on protein-coding genes.
2) Physical Ribosomal RNA (rRNA) removal
Ribosomal rRNA can also be removed by hybridization to complementary biotinylated oligo probes, followed by extraction with streptavidin-coated magnetic beads. riboPOOLs developed by siTOOLs Biotech efficiently removes rRNA through this route, with a workflow similar to Ribo-Zero from Illumina.
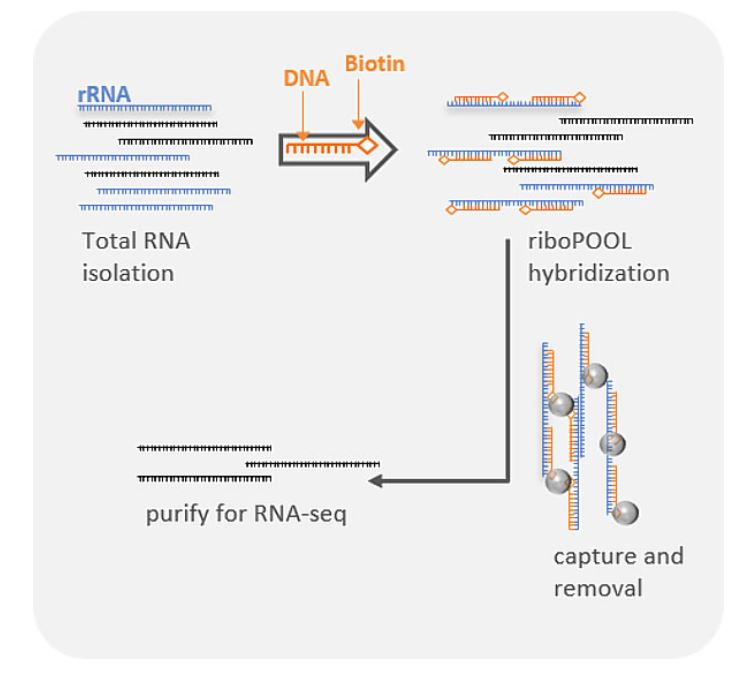
Compared to polyA selection methods, rRNA removal enables detection of non-polyadenylated transcripts and small RNAs. Comparisons between differential gene expression detected with both methods were typically well-correlated. The rRNA removal method however could detect both long and short transcripts showing less of a 3′ bias than polyA selection.
Physical rRNA removal also performs better for degraded and FFPE samples, and can also be applied for metagenomic samples that contain microbes. The Pan-Prokaryote riboPOOL by siTOOLs for example, functions effectively to remove rRNA from a diverse range of prokaryotic species, and can be used in combination with human and mouse/rat riboPOOLs to deplete rRNA from complex samples containing multiple species.


By using targeted probes, one can also flexibly deplete abundant RNAs that take up expensive RNA-Seq reads. For example, globin, found in high amounts in RNA isolated from blood samples, can be efficiently depleted by globin mRNA-specific probes.
Ribosomal RNA can also be removed by selective degradation where enzyme RNase H is used to specifically degrade DNA-RNA hybrids formed between DNA probes and complementary rRNA (e.g. NEBNext rRNA depletion kit by New England Biolabs). This method was reported to produce consistent results, working as well on degraded samples though there was a bias against detecting transcripts of shorter length compared to Ribo-Zero.
3) Targeted amplification
An alternative method to deplete rRNA involves the use of “not so random” hexamer/heptamer primers with a decreased affinity for rRNA during first strand cDNA synthesis. This is employed by the Ovation RNA-Seq kits from NuGen. Though the kit can be used to detect non-polyA RNAs and can be applied to prokaryotes, the additional incorporation of oligo(dT) still contributes to a bias towards detecting 3′ regions.
A recent ribosome profiling study comparing library preparation methods reported fewer reads obtained and greater intronic reads for Nugen kits compared to polyA-selection methods. As Nugen also incorporated an RNase-mediated degradation of unwanted transcripts during final library construction steps, this indicates targeted amplification alone cannot totally remove rRNA. The method does however work with low input amounts and degraded samples.

So which ribo-depletion method works best?
And the answer as always? It depends. Depending on the ribo-depletion method chosen in RNA-Seq library preparation, some differences in genes detected and their expression levels will certainly be observed.
Poly-A selection might be the most efficient method when only focussed on protein-coding genes, but one loses significant information on non-polyadenylated RNAs and immature transcripts. In instances such as microbial sequencing or in sequencing degraded or FFPE samples, poly-A selection cannot even be applied or may perform poorly.
Physical rRNA removal offers the advantage of retrieving more transcriptomic information but comes at a cost of greater intronic/intergenic reads that necessitates a greater sequencing depth. However, it offers greater flexibility and better performance in sequencing of challenging sample types.
Targeted amplification with “not so random” primers though effective for low input material, comes also at a cost of greater sequencing depth required.
All methods are subject to some extent of non-specificity and detection bias. Further variability can also arise from different methods of sequence alignment in RNA-Seq data analysis. It is therefore always advisable to validate sequencing data obtained by real-time quantitative PCR (rtqPCR) or other methods.
References:
1. Song, Y., Milon, B., Ott, S., Zhao, X., Sadzewicz, L., Shetty, A., Boger, E. T., Tallon, L. J., Morell, R. J., Mahurkar, A., and Hertzano, R. (2018) A comparative analysis of library prep approaches for sequencing low input translatome samples. BMC Genomics. 19, 696
2. O’Neil, D., Glowatz, H., and Schlumpberger, M. (2013) Ribosomal RNA Depletion for Efficient Use of RNA-Seq Capacity. in Current Protocols in Molecular Biology, p. 4.19.1-4.19.8, John Wiley & Sons, Inc., Hoboken, NJ, USA, 103, 4.19.1-4.19.8
3. Stark, R., Grzelak, M., and Hadfield, J. (2019) RNA sequencing: the teenage years. Nat. Rev. Genet. 10.1038/s41576-019-0150-2
4. Cui, P., Lin, Q., Ding, F., Xin, C., Gong, W., Zhang, L., Geng, J., Zhang, B., Yu, X., Yang, J., Hu, S., and Yu, J. (2010) A comparison between ribo-minus RNA-sequencing and polyA-selected RNA-sequencing. Genomics. 96, 259–265
5. Zhao, S., Zhang, Y., Gamini, R., Zhang, B., and von Schack, D. (2018) Evaluation of two main RNA-seq approaches for gene quantification in clinical RNA sequencing: polyA+ selection versus rRNA depletion. Sci. Rep. 8, 4781
6. Herbert, Z. T., Kershner, J. P., Butty, V. L., Thimmapuram, J., Choudhari, S., Alekseyev, Y. O., Fan, J., Podnar, J. W., Wilcox, E., Gipson, J., Gillaspy, A., Jepsen, K., BonDurant, S. S., Morris, K., Berkeley, M., LeClerc, A., Simpson, S. D., Sommerville, G., Grimmett, L., Adams, M., and Levine, S. S. (2018) Cross-site comparison of ribosomal depletion kits for Illumina RNAseq library construction. BMC Genomics. 19, 199
Featured Image is an artist’s rendition of a ribosome. Credit: C. BICKLE/SCIENCE













{kind=link}