
Correcting seed-based off-target effects in RNAi screens
Summary: Correcting for seed-based off-targets can improve the results from RNAi screening. However, the correlation between siRNAs for the same gene is still poor and the strongest screening hits remain difficult to interpret.
Seed-based off-target correction has little effect on reagent reproducibility
Given that seed-based off-targets are the main cause of phenotypes in RNAi screening, trying to correct for those effects makes good sense.
The dominance of seed-based off-targets means that independent siRNAs for the same gene usually show poor correlation.
If one could correct for the seed effect, the correlation between siRNAs targeting the same gene may improve.
One straightforward way to do seed correction is to subtract the ‘seed median’ from each siRNA. (The seed median is the median for all siRNAs having the given seed.)
This was the approach used by Grohar et al. in a recent genome-wide survey of EWS-FLI1 splicing (involved in Ewing sarcoma). They used the Silencer Select library, which has 3 siRNAs per target gene.
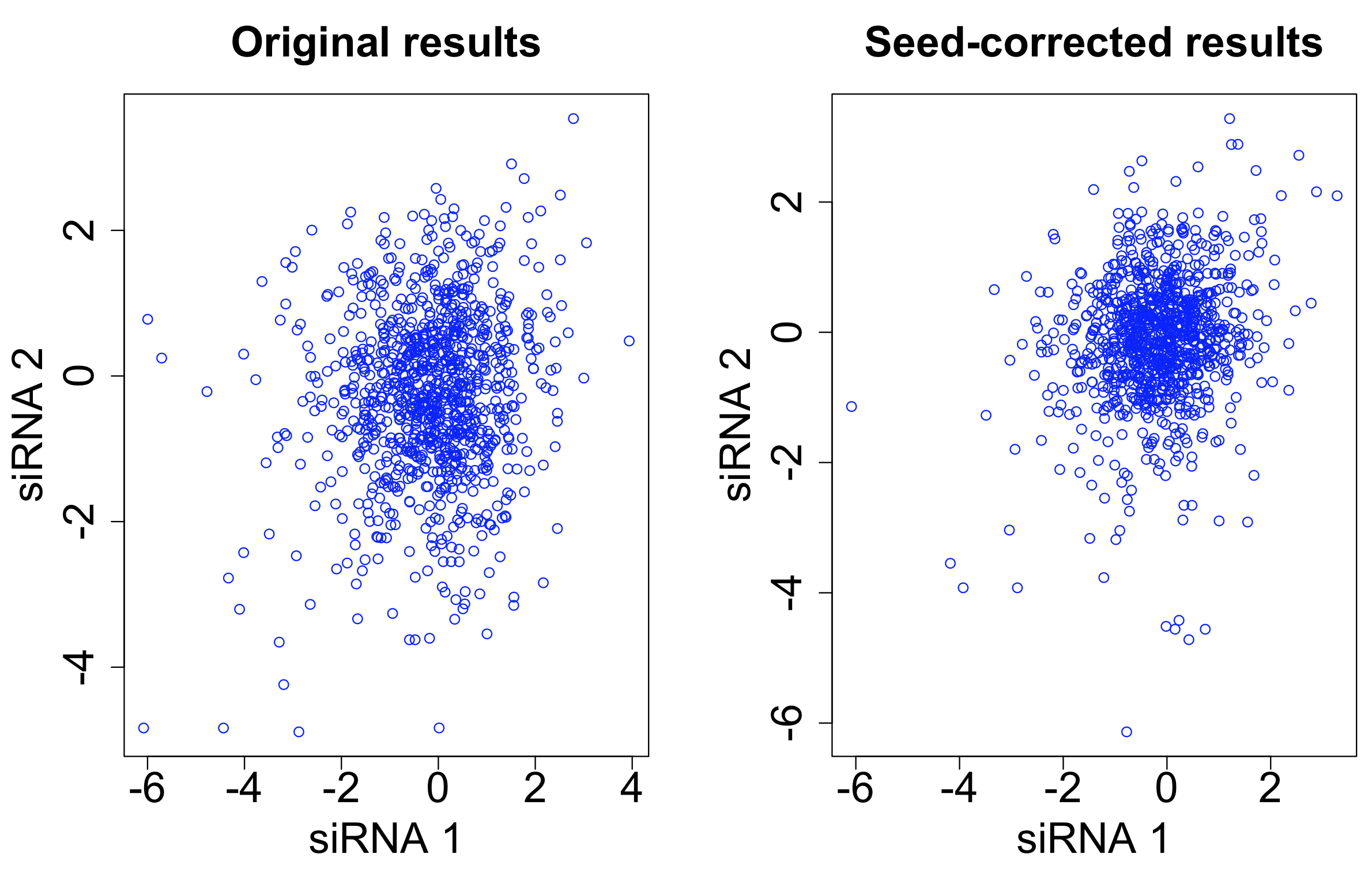
After seed correction, there is only minor improvement in the correlation between siRNAs targeting the same gene. The intra-class correlation (ICC) improves from 0.031 to 0.037. The ICC for siRNAs with the same 7-mer seed decreases from 0.576 to 0.261.
Although we have reduced the seed-based signal, it has not resulted in a correspondingly large improvement in the gene-based signal.
More sophisticated seed correction can improve reagent correlation
Grohar et al. used a simple seed-median subtraction method to correct their screening results.
A more sophisticated method (scsR) was developed by Franceshini et al. for seed-based correction of screening data. It corrects using the mean value for siRNAs with the same seed, and weighs the correction using the standard deviation the values. This allows seeds with a more consistent effect to contribute more to the data normalisation.
Applying the scsR method to the Grohar data, ICC for siRNAs targeting the same gene increases from 0.031 to 0.041. It is better than the increase with seed-median subtraction (0.037), but is still only a fairly minor improvement (plot created using random selection of 10,000 pairs of siRNAs that target the same gene):
Off-target correction increases double-hit rate in top siRNAs of RNAi screen
The following plot shows the count for single-hit and double-hit genes as we go through the top 1000 siRNAs (of ~60K screened in total). Double-hit means that the gene is covered by 2 (or more) hit siRNAs.
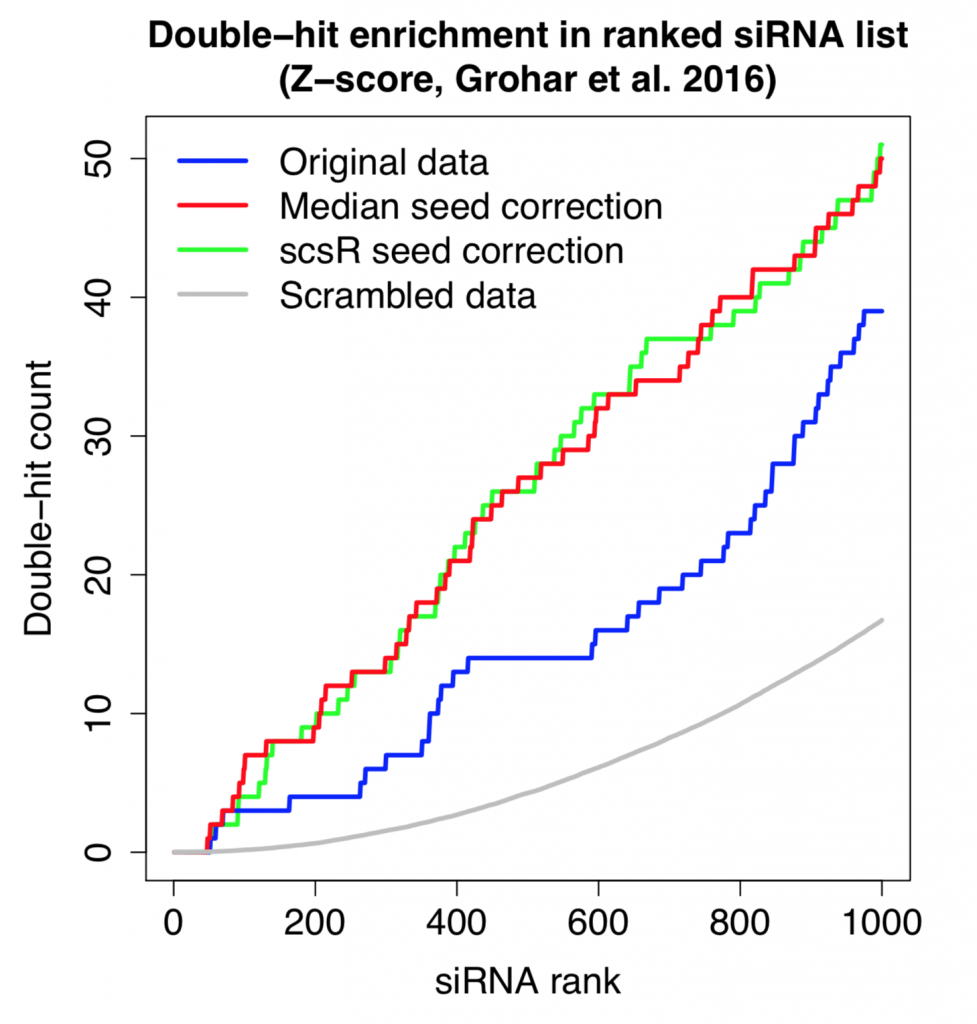
Despite the small improvement in reagent correlation, the double-hit rate is essentially the same using simple seed-median subtraction or the more advanced scsR method.
Furthermore, the number of double-hits is higher than what we’d expect by chance.
This shows that, despite the noise from off-target effects, there is some on-target signal that can be detected.
siRNAs with the strongest phenotypes remain difficult to interpret
Despite the fact that the double-hit count is higher than expected by change, most of the genes targeted by the strongest siRNAs are single-hits. siRNAs with the strongest phenotypes remain difficult to interpret.

Seed correction is best suited for single-siRNA libraries. Low-complexity pools, like siGENOME or ON-TARGETplus, are less amenable to effective seed correction since there are (usually) 4 different seeds per pool. This reduces the effectiveness of seed-based correction, even though seed-based off-target effects remain the primary determinant of observed phenotypes (as discussed here, here , and here).
The best way to correct for seed-based off-targets is to avoid them in the first place. Using more specific reagents, like high-complexity siPOOLs, is the key to generating interpretable RNAi screening results.
For help with seed correction or other RNAi screening data analysis with the Phenovault, contact us at info@sitools.de